Research Articles
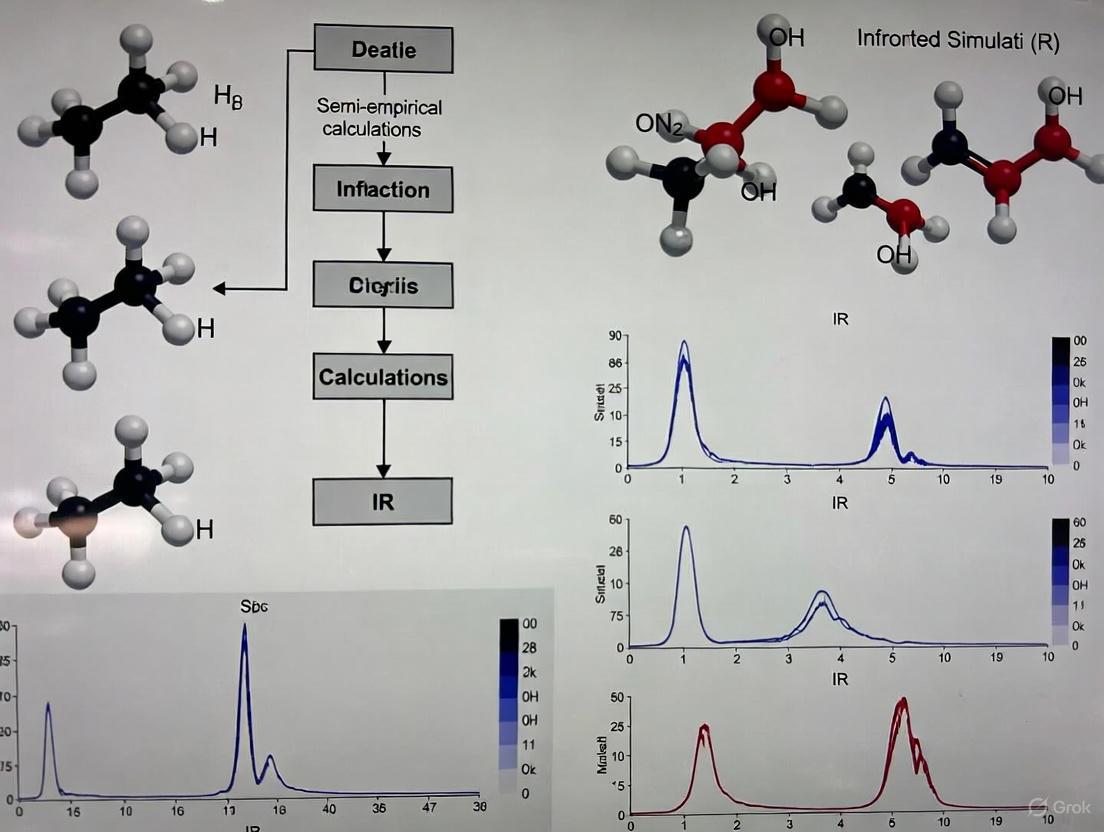
Semi-Empirical Methods for IR Spectra Simulation: A Practical Guide for Biomedical Researchers
This comprehensive review explores semi-empirical quantum mechanical methods for simulating infrared (IR) spectra, addressing critical needs in drug discovery and materials science.
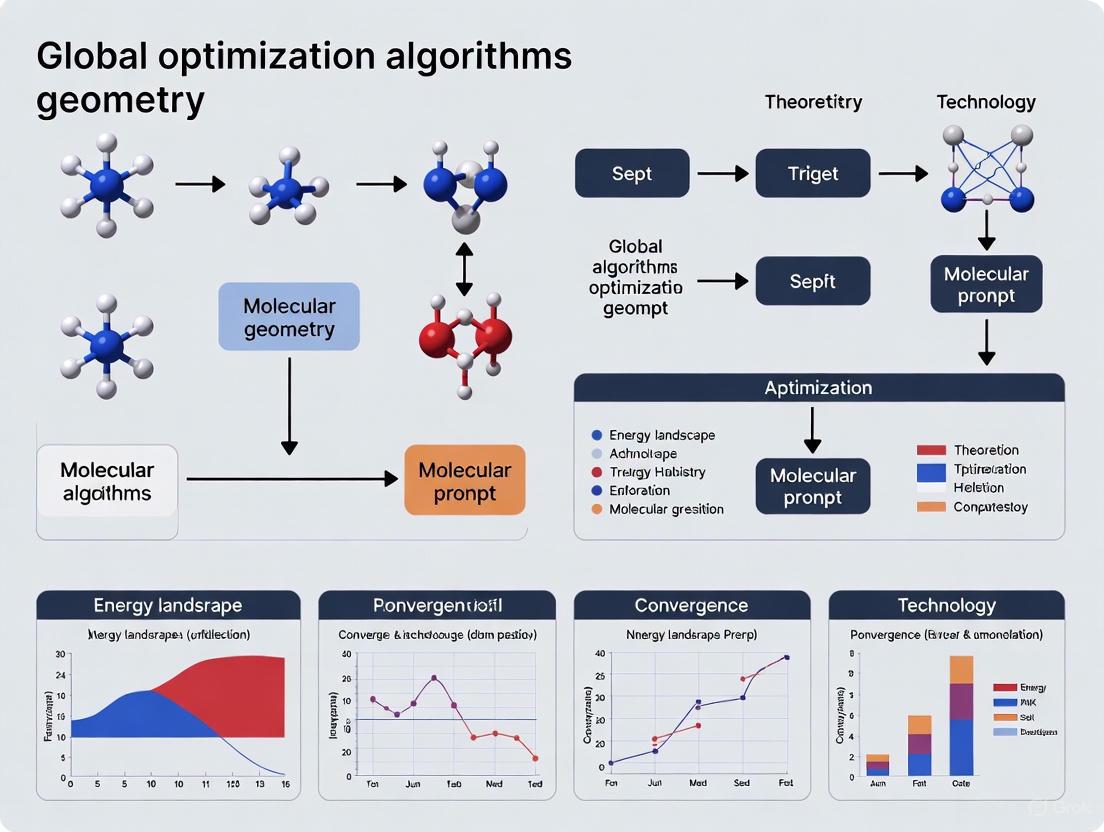
Global Optimization Algorithms for Molecular Geometry: From Fundamentals to AI-Driven Drug Discovery
This article provides a comprehensive overview of global optimization algorithms for predicting molecular geometries, a critical task in computational chemistry and drug discovery.
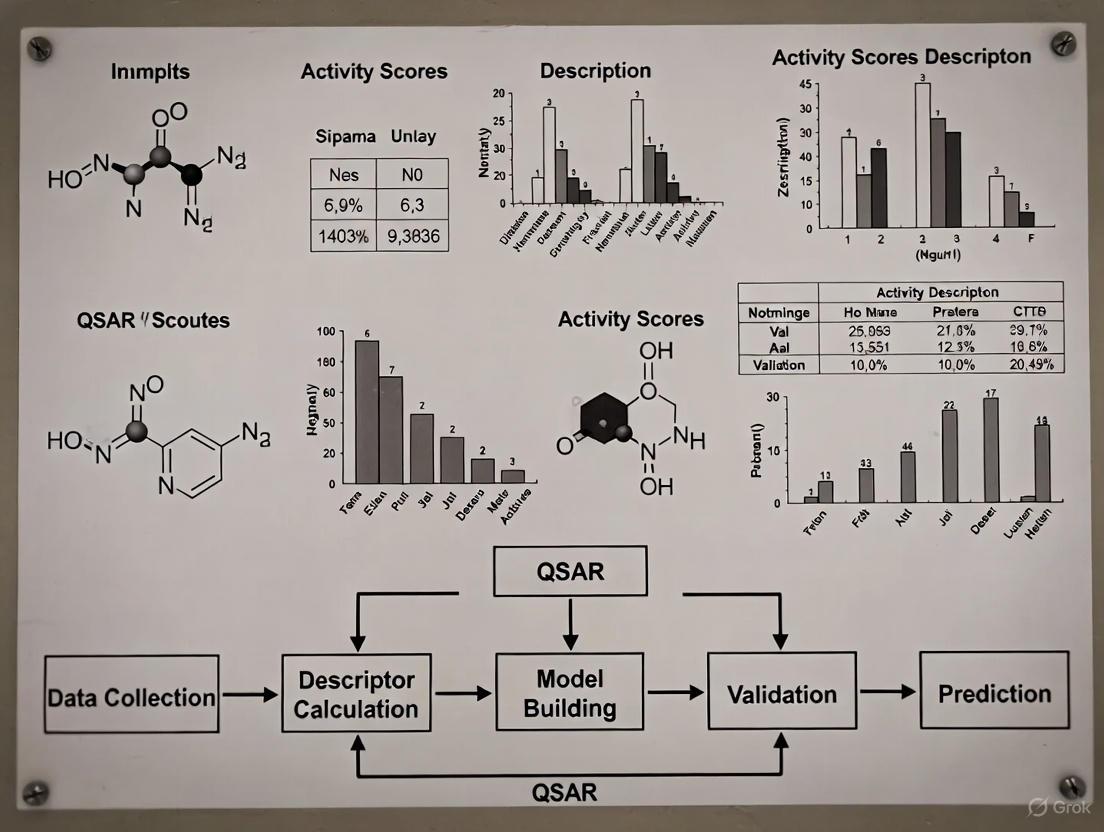
QSAR Techniques in Modern Drug Discovery: From Foundational Principles to AI-Driven Applications
This article provides a comprehensive overview of Quantitative Structure-Activity Relationship (QSAR) modeling, a cornerstone computational method in chemical and pharmaceutical research.
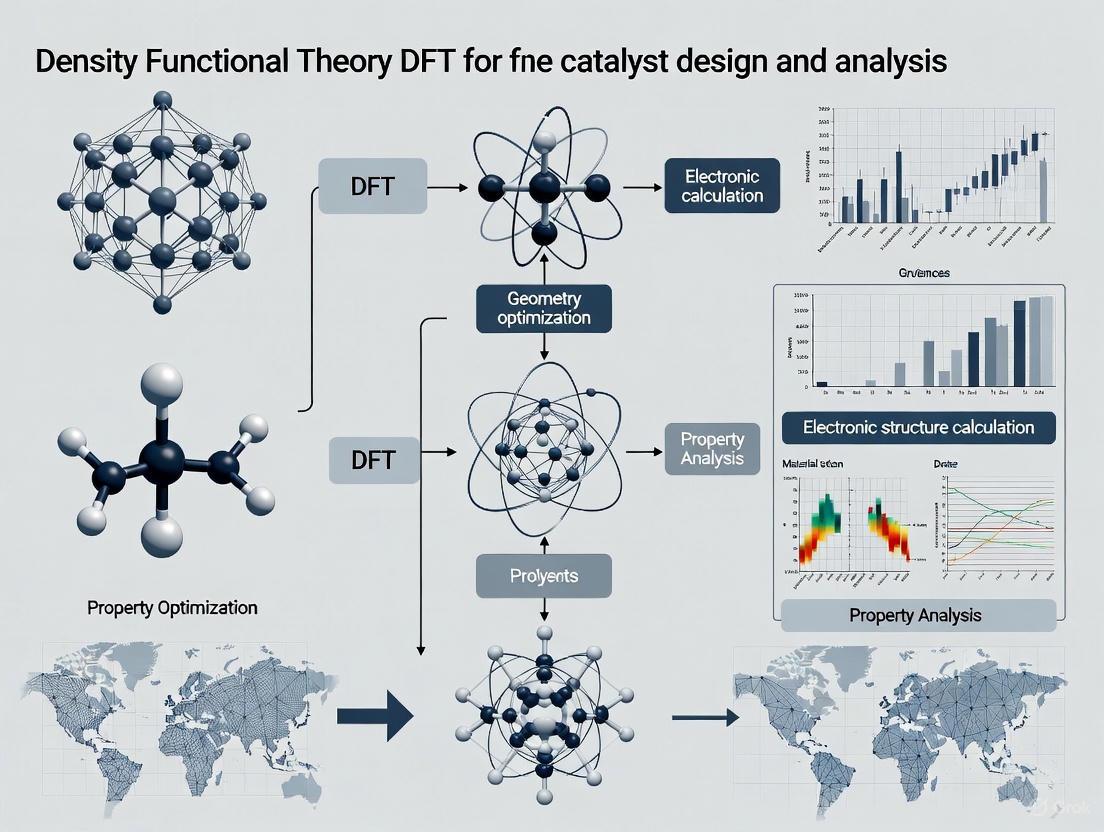
Density Functional Theory for Catalyst Design: From Fundamentals to AI-Driven Discovery
This article provides a comprehensive overview of the application of Density Functional Theory (DFT) in catalyst design and analysis, tailored for researchers and development professionals.
Machine Learning in Homogeneous Catalysis: A Comprehensive Guide to Data-Driven Optimization and Design
This article provides a comprehensive overview of the transformative role of machine learning (ML) in optimizing homogeneous transition-metal catalysis, a cornerstone of modern synthetic chemistry and pharmaceutical development.
Protein Folding Unraveled: How Molecular Dynamics Simulations Guide Biomolecular Discovery and Drug Design
This article provides a comprehensive overview of the application of molecular dynamics (MD) simulations in protein folding studies, tailored for researchers and drug development professionals.
Quantum Mechanical Methods for Molecular Structure Prediction: From Fundamentals to Drug Discovery Applications
This article provides a comprehensive review of quantum mechanical (QM) methods for molecular structure prediction, tailored for researchers and drug development professionals.
Bayesian Models for Chemical Probe Quality Prediction: Advancing Drug Discovery with Machine Learning
This article explores the transformative role of Bayesian statistical models in predicting the quality and viability of chemical probes for drug discovery.
From Theory to Therapy: How the Schrödinger Equation Powers Modern Drug Discovery
This article explores the pivotal role of the Schrödinger equation in advancing computational chemistry and drug discovery.
From Quantum Calculations to Complex Systems: How the 1998 and 2013 Nobel Prizes Revolutionized Computational Chemistry
This article explores the foundational breakthroughs of the 1998 and 2013 Nobel Prizes in Chemistry, which established computational methods as a cornerstone of modern chemical research.