A Practical Guide to Molecular Property Prediction with Machine Learning for Drug Discovery
This article provides a comprehensive roadmap for researchers and drug development professionals to implement machine learning for molecular property prediction.
A Practical Guide to Molecular Property Prediction with Machine Learning for Drug Discovery
Abstract
This article provides a comprehensive roadmap for researchers and drug development professionals to implement machine learning for molecular property prediction. It covers the foundational concepts of molecular representations, explores predictive and generative modeling methodologies, addresses critical troubleshooting and optimization challenges like dataset bias and model uncertainty, and outlines rigorous validation and comparative analysis techniques. The guide synthesizes current best practices to help scientists navigate the complexities of AI-driven drug discovery, from initial setup to reliable deployment, with a focus on practical application and building confidence in predictive outcomes.
Laying the Groundwork: Core Concepts and Molecular Representations for ML
The Challenge of Navigating Vast Chemical Space
The chemical space of possible, stable organic molecules is estimated to encompass 10^60 to 10^100 distinct structures, a scale so vast that exhaustive experimental characterization is an impossibility. This presents a fundamental challenge for scientific research and industrial development in fields like drug discovery and materials science. The process of experimentally determining molecular properties is notoriously slow, expensive, and resource-intensive, often requiring sophisticated equipment and consuming significant time. For instance, the traditional drug discovery process can take over a decade and cost billions of dollars, with a success rate of only 1 in 5,000 compounds [1].
Machine learning (ML) has emerged as a transformative tool to navigate this expansive space. By learning from existing chemical data, ML models can predict the properties of new, unsynthesized molecules with high speed and accuracy, dramatically accelerating the search for new drugs, materials, and energy carriers. However, the effectiveness of these models is constrained by several significant challenges, including the scarcity of high-quality experimental data, the difficulty of generalizing predictions to new regions of chemical space, and the complexity of selecting the appropriate model architecture and featurization for a given task [2] [3] [4]. This guide provides a technical overview for researchers and scientists embarking on ML-driven molecular property prediction, with a focus on overcoming these central challenges.
Core Machine Learning Approaches
Molecular property prediction leverages several classes of ML algorithms, each with distinct strengths and methodological considerations. The selection of an approach often depends on the volume of available data and the specific prediction task.
Multi-Task Learning (MTL) and the Negative Transfer Problem
MTL is a powerful strategy designed to alleviate data scarcity by training a single model on multiple related property prediction tasks simultaneously. The core idea is that by learning these tasks jointly, the model can discover and exploit shared underlying structures and correlations, leading to improved generalization on all tasks [2].
- Architecture: A typical MTL model consists of a shared backbone (e.g., a Graph Neural Network) that learns a general-purpose molecular representation, and multiple task-specific heads (e.g., Multi-Layer Perceptrons) that map the shared representation to a specific property value [2].
- Methodology: The model is trained on a dataset where each molecule has labels for one or more properties. The total loss is often a weighted sum of the losses for each individual task.
- Challenge - Negative Transfer (NT): A major obstacle to effective MTL is negative transfer, which occurs when updates driven by one task are detrimental to the performance of another. This is often exacerbated by task imbalance, where certain tasks have far fewer labeled examples than others, limiting their influence on the shared model parameters [2].
- Advanced Protocol: Adaptive Checkpointing with Specialization (ACS)
- Purpose: To mitigate negative transfer while preserving the benefits of inductive transfer [2].
- Procedure:
- Train a single MTL model with a shared GNN backbone and task-specific MLP heads.
- Throughout training, continuously monitor the validation loss for every single task.
- Independently for each task, checkpoint and save the model parameters (both the shared backbone and the task-specific head) whenever that task's validation loss reaches a new minimum.
- Upon completion, each task has a specialized model that represents the optimal shared knowledge for that specific property.
- Validation: This method has been shown to consistently surpass or match the performance of recent supervised methods, particularly in ultra-low data regimes. In one real-world application, it enabled accurate predictions for sustainable aviation fuel properties with as few as 29 labeled samples [2].
Geometric Deep Learning
Geometric deep learning incorporates three-dimensional structural information into the model, which is crucial for accurately predicting properties that depend on molecular conformation and quantum-chemical interactions.
- Architecture: The Directed Message Passing Neural Network (D-MPNN) is a state-of-the-art architecture for molecular graphs [4]. Messages are passed along directed edges (bonds) to prevent redundant updates and noise. A Geometric D-MPNN extends this by incorporating 3D molecular coordinates and other quantum-chemical descriptors into the featurization of nodes (atoms) and edges [4].
- Featurization: Models can be featurized with 2D information (molecular graph) or 3D information (atomic coordinates from methods like Density Functional Theory - DFT). The necessity of 3D information varies with the property being predicted [4].
- Advanced Protocol: Achieving Chemical Accuracy with Δ-ML and Transfer Learning
- Chemical Accuracy: A key target, especially for thermochemistry, is an error of ~1 kcal mol⁻¹, which is required for constructing thermodynamically consistent models [4].
- Δ-ML Protocol:
- Obtain property calculations for a set of molecules at both a low (LL) and high (HL) level of theory.
- Train an ML model to predict the residual (difference) between the HL and LL values:
Δ = Property_HL - Property_LL. - The final prediction for a new molecule is the sum of the easily computed low-level value and the ML-predicted delta:
Property_HL_Predicted = Property_LL + Δ_Predicted[4].
- Transfer Learning Protocol:
- Pre-training: A model is first trained on a large, diverse database of molecules with properties calculated at a lower level of theory (e.g., via COSMO-RS). This teaches the model a robust general molecular representation.
- Fine-tuning: The pre-trained model is then further trained (fine-tuned) for a few epochs on a small, high-quality dataset (e.g., experimental data or high-level quantum chemical data) for the specific property of interest. This adapts the model to achieve high accuracy on the target domain [4].
Addressing the Out-of-Distribution (OOD) Generalization Challenge
A model's ability to make accurate predictions for molecules that are structurally different from those in its training set—known as OOD generalization—is critical for real-world discovery, which inherently involves exploring new chemical territories.
- The Problem: Standard ML models often suffer from significant performance degradation when applied to OOD molecules. The BOOM benchmark study found that even top-performing models exhibited an average OOD error three times larger than their in-distribution error [3].
- Benchmarking Insights: The BOOM benchmark evaluated over 140 model and task combinations, revealing that no single existing model achieves strong OOD generalization across all tasks. Key findings include [3]:
- Models with high inductive bias (e.g., strong architectural constraints suited to the problem) can perform well on OOD tasks with simple, specific properties.
- Current large-scale chemical foundation models, while promising for limited-data scenarios, do not yet show strong OOD extrapolation capabilities.
- Methodological Recommendations: To enhance OOD performance, the benchmark emphasizes careful attention to data generation procedures, model pre-training strategies, hyperparameter optimization, and molecular representation [3].
Experimental Protocols and Workflows
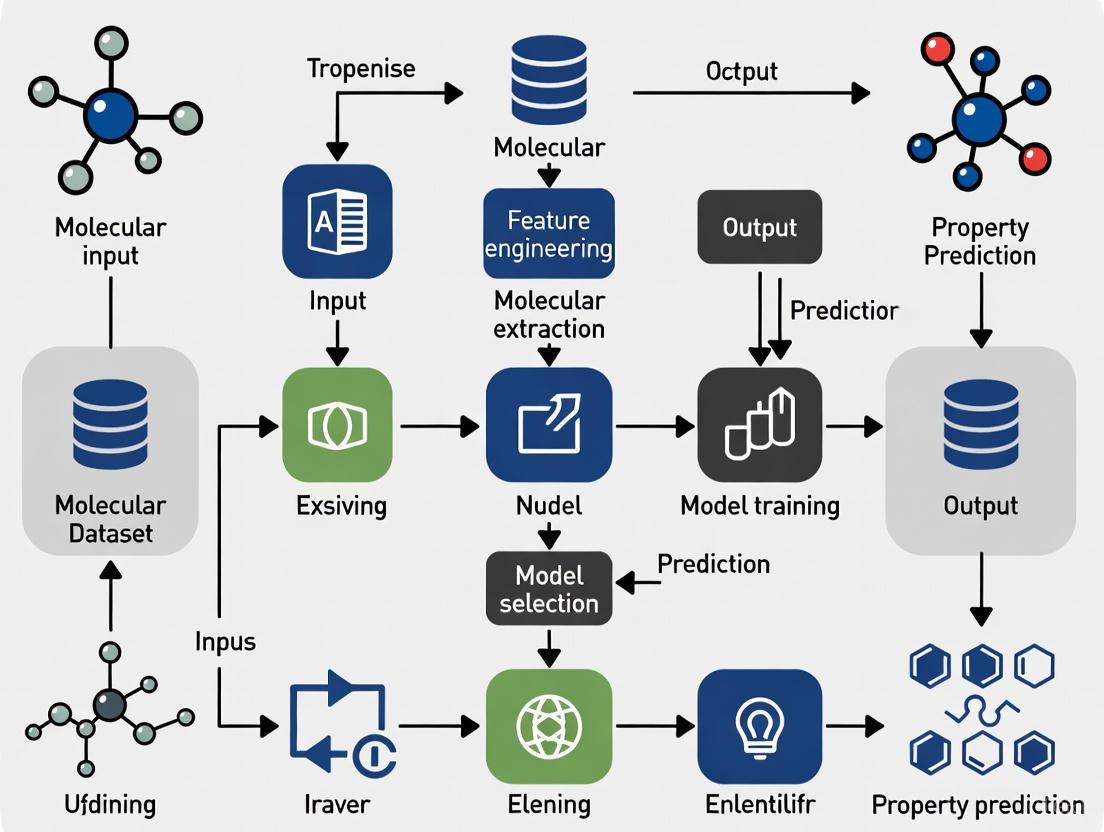
Implementing a robust ML pipeline for property prediction requires a structured workflow from data preparation to model deployment.
A Standard Workflow for Molecular Property Prediction
The following diagram illustrates a generalized experimental protocol that incorporates best practices for handling data splits and specialized training techniques.
Data Splitting Methodologies
The strategy for splitting data into training, validation, and test sets is critical for a realistic performance estimate.
- Random Splitting: Divides molecules randomly. This often leads to over-optimistic performance estimates because molecules in the test set can be highly structurally similar to those in the training set [2] [3].
- Scaffold Splitting (Murcko Scaffold): Partitions molecules based on their Bemis-Murcko scaffold (the core ring system with linkers). This ensures that molecules with different core structures are in different splits, providing a more challenging and realistic assessment of a model's ability to generalize to novel chemotypes [2].
- Temporal Splitting: Splits data based on the year of measurement or publication. This best simulates a real-world discovery scenario where models are used to predict properties for molecules synthesized in the future. Studies have shown that temporal splits can reveal inflated performance estimates from random/scaffold splits [2].
Successful ML projects rely on a suite of software tools, datasets, and computational resources. The table below summarizes key "research reagent solutions" for molecular property prediction.
Table 1: Essential Tools and Resources for Molecular Property Prediction
| Item Name | Type | Primary Function | Key Features / Considerations |
|---|---|---|---|
| Directed MPNN (D-MPNN) [2] [4] | Model Architecture | Graph-based property prediction | Reduces redundant messaging; supports 2D and 3D featurization. |
| ACS Training Scheme [2] | Training Algorithm | Mitigates negative transfer in MTL | Adaptive checkpointing for task specialization; ideal for imbalanced data. |
| Δ-ML & Transfer Learning [4] | Training Protocol | Achieves high accuracy with limited data | Δ-ML corrects low-level calculations; transfer learning adapts from large pretraining sets. |
| Therapeutics Data Commons (TDC) [1] | Data Resource | Benchmark datasets for drug discovery | Provides curated datasets for various drug development tasks. |
| BOOM Benchmark [3] | Benchmarking Tool | Evaluates out-of-distribution (OOD) generalization | Systematically tests model performance on OOD molecules. |
| ChemXploreML [5] | Software Application | User-friendly desktop app for predictions | No programming skills required; operates offline; uses molecular embedders. |
| RDKit [6] | Software Library | Open-source cheminformatics | Fundamental for molecule handling, fingerprint generation, and descriptor calculation. |
| Geometric Datasets (ThermoG3, DrugLib36) [4] | Data Resource | Large-scale quantum chemical data | Provides high-quality, industrially-relevant data for training and pretraining. |
Quantitative Performance Benchmarks
To guide model selection and expectation setting, it is essential to consider typical performance metrics across different model types and data regimes.
Table 2: Performance Benchmarks of ML Models on Molecular Property Prediction
| Model / Approach | Dataset / Context | Key Performance Metric | Result / Accuracy | Notes / Implication |
|---|---|---|---|---|
| ACS (MTL) [2] | ClinTox, SIDER, Tox21 | Average Improvement vs. Single-Task Learning | +8.3% improvement | Demonstrates effectiveness of transfer and NT mitigation. |
| Geometric D-MPNN [4] | Novel Thermochemistry Data | Mean Absolute Error (MAE) | Achieved <1 kcal mol⁻¹ ("Chemical Accuracy") | Crucial for reliable thermochemical kinetic models. |
| Schrödinger Formulation ML [7] | Polymer Glass Transition (Tg) | Coefficient of Determination (R²) | R² = 0.97 | High accuracy for complex mixture properties. |
| ChemXploreML App [5] | Critical Temperature Prediction | Accuracy Score | Up to 93% accuracy | Validates use of accessible, non-programmatic tools. |
| Top BOOM Benchmark Model [3] | Various OOD Tasks | OOD vs. In-Distribution Error | OOD error 3x larger than ID error | Highlights the pervasive challenge of OOD generalization. |
| ACS (MTL) [2] | Sustainable Aviation Fuels | Minimum Viable Dataset Size | Accurate models with 29 samples | Enables work in ultra-low data regimes. |
Machine learning provides a powerful and necessary set of tools for navigating the vastness of chemical space. The field has moved beyond simple models to sophisticated frameworks that can handle multi-task learning, 3D geometric information, and specialized training protocols like ACS and Δ-ML to achieve chemical accuracy. However, significant challenges remain. Out-of-distribution generalization is the next frontier, with current benchmarks showing that even the best models struggle to reliably extrapolate [3]. Future progress will likely come from models with stronger inductive biases, improved pre-training strategies on large, diverse datasets, and more rigorous evaluation protocols that prioritize real-world generalization over performance on narrow benchmarks. By leveraging the tools, protocols, and insights summarized in this guide, researchers can effectively harness ML to accelerate the discovery of new molecules with tailored properties.
Key Computational Representations of Molecules
The acceleration of materials discovery and drug development increasingly hinges on the ability of machine learning (ML) models to accurately predict molecular properties. The foundation of any successful ML model in this domain is its computational molecular representation, which translates chemical structures into a format that algorithms can process. The choice of representation fundamentally shapes the model's capacity to capture the physical and chemical principles that govern molecular behavior.
This guide provides a technical overview of the core representations used in modern ML-driven molecular property prediction, detailing their methodologies, comparative advantages, and protocols for implementation.
Molecular Graph Representations
Molecular graphs provide a natural and intuitive framework for representing molecules by treating atoms as nodes and chemical bonds as edges. This structure is inherently compatible with Graph Neural Networks (GNNs), which learn through a process of message passing [8] [9].
Core Methodology: Message Passing Neural Networks (MPNNs)
The message-passing framework allows atoms (nodes) to aggregate information from their local chemical environments. The process can be formalized in three key steps [8]:
- Message (M): For each node (atom)
v, a message is constructed from its neighboring nodesuand the connecting edges (bonds). This function,M, typically combines the current state of the neighborh_uand the features of the bonde_uv. - Aggregation (AGG): The messages from all neighbors are aggregated into a single vector. Common aggregation functions include sum, mean, or maximum.
- Update (U): The central node's state
h_vis updated based on its previous state and the aggregated message from its neighbors.
This cycle is repeated for multiple steps, allowing each atom to incorporate information from atoms increasingly farther away in the molecular structure, effectively capturing the topological environment [9].
Diagram 1: The Message Passing Neural Network (MPNN) workflow for learning molecular representations from graphs.
Experimental Protocol: Implementing a GNN for Property Prediction
A standard protocol for training a GNN on molecular property prediction involves the following steps [8]:
- Data Preparation: A dataset such as MoleculeNet is used. Molecules are converted into graphs, where nodes are featurized with atomic properties (e.g., atom type, degree, hybridization) and edges are featurized with bond properties (e.g., bond type, conjugation).
- Model Architecture: A GNN model like a Graph Convolutional Network (GCN) or Graph Attention Network (GAT) is constructed. The model consists of several message-passing layers followed by a readout function.
- Readout / Pooling: After the final message-passing layer, the updated node states are combined to form a single graph-level representation. This is often done via global mean pooling, sum pooling, or more advanced attention-based methods.
- Prediction Head: The graph-level representation is passed through a fully connected neural network to produce the final property prediction (e.g., solubility, toxicity).
- Training: The model is trained using a relevant loss function (e.g., Mean Squared Error for regression, Cross-Entropy for classification) and an optimizer like Adam.
Geometric and 3D Representations
While 2D graph representations capture topological connectivity, they ignore the crucial three-dimensional spatial arrangement of atoms. Geometric representations are essential for predicting properties that depend on molecular conformation, such as energy, vibrational spectra, and reactivity [10] [11].
Density Functional Theory (DFT) as a Data Source
Geometric ML models are typically trained on data generated from high-fidelity quantum mechanical calculations, primarily Dispersion-inclusive Density Functional Theory (DFT) [10] [11]. DFT provides the ground-truth labels for properties like system energy and atomic forces. Large-scale DFT datasets, such as the Open Molecules 2025 (OMol25) dataset, provide the foundational data for training these models [12] [11].
OMol25 is a collection of over 100 million DFT calculations on systems containing up to 350 atoms, spanning 83 elements and a wide range of chemical interactions [11]. This data is used to train Machine Learning Interatomic Potentials (MLIPs), which can predict DFT-level accuracy at a fraction of the computational cost, enabling simulations of large-scale systems that are otherwise prohibitive [12].
Advanced Architectural Innovation: Kolmogorov-Arnold GNNs (KA-GNNs)
Recent research has explored enhancing GNNs by integrating them with Kolmogorov-Arnold Networks (KANs). KA-GNNs replace the standard linear transformations and fixed activation functions in traditional GNNs with learnable univariate functions placed on the edges of the network [13].
The methodology involves integrating KAN modules into the three core components of a GNN [13]:
- Node Embedding: KANs are used to initialize node features from atomic and local environmental data.
- Message Passing: The aggregation and update functions within the GNN are handled by KAN layers, which use Fourier-series-based basis functions to capture complex patterns.
- Readout: The final pooling step to a graph-level representation is also performed by a KAN.
This integration has been shown to improve both prediction accuracy and computational efficiency while offering enhanced interpretability by highlighting chemically meaningful substructures [13].
Emerging and Multimodal Representations
Functional Group-Level Reasoning with LLMs
For large language models (LLMs), a key representation is the functional group-level annotation. Benchmarks like FGBench provide datasets where functional groups (e.g., hydroxyl, carbonyl) are precisely annotated and localized within the molecule [14]. This representation provides valuable prior knowledge that links molecular structures with textual descriptions, allowing LLMs to reason about the impact of specific chemical moieties on overall molecular properties [14].
Multimodal Representations: Integrating Text and Geometry
Another emerging approach is multimodal learning, which enriches geometric graph representations with textual descriptors from public databases like PubChem. These descriptors can include IUPAC names, molecular formulas, and physicochemical properties [15].
The experimental protocol for this approach involves [15]:
- Feature Extraction: Separate feature vectors are generated for the molecular graph (using a GNN) and for the textual descriptors (using an encoder network).
- Gated Fusion: A gated fusion mechanism dynamically balances and combines the geometric and textual feature vectors into a unified representation. This allows the model to leverage complementary information from both data types.
Comparative Analysis of Molecular Representations
The table below summarizes the key characteristics, strengths, and limitations of the primary computational representations.
Table 1: Comparative Analysis of Key Molecular Representations
| Representation | Core Idea | Best For | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Molecular Graph (GNN) [8] [9] | Atoms as nodes, bonds as edges. | Predicting properties dependent on 2D topology (e.g., toxicity, drug-likeness). | Natural representation; automatically learns features; invariant to atom indexing. | Neglects 3D spatial and electronic structure. |
| Geometric (3D) Representation [10] [11] | Includes 3D atomic coordinates. | Energetics, forces, spectroscopy, and conformation-dependent properties. | Captulates essential physics; enables accurate force fields (MLIPs). | Computationally intensive; requires high-quality 3D data. |
| KAN-enhanced Graph (KA-GNN) [13] | GNNs with learnable activation functions on edges. | General molecular property prediction with enhanced accuracy/interpretability. | Improved parameter efficiency and interpretability; strong theoretical foundation. | Emerging technology; less established than traditional GNNs. |
| Functional-Group (LLM) [14] | Textual annotation of chemical substructures. | Reasoning about structure-property relationships in language models. | Provides chemical prior knowledge; interpretable; leverages LLM capabilities. | Struggles with precise quantitative prediction; model may hallucinate. |
| Multimodal [15] | Combines graph and textual descriptors. | Leveraging existing chemical metadata to boost prediction on benchmarks. | Enriches representation with diverse data sources; can improve performance. | Gains can be task-dependent; adds model complexity. |
Table 2: Key datasets, models, and tools for molecular property prediction research.
| Resource Name | Type | Key Features | Primary Use Case |
|---|---|---|---|
| OMol25 Dataset [11] [16] | Dataset | >100M DFT calculations, 83 elements, up to 350 atoms, electronic densities/wavefunctions. | Training next-generation MLIPs and electronic property prediction models. |
| OMC25 Dataset [10] | Dataset | ~27M molecular crystal structures from DFT relaxation trajectories. | Developing models for crystalline materials and solid-state properties. |
| FGBench [14] | Dataset & Benchmark | 625K problems with functional-group annotations for reasoning. | Training and evaluating LLMs on fine-grained molecular reasoning tasks. |
| MoleculeNet [8] [2] | Benchmark Suite | Curated collection of datasets for molecular property prediction. | Standardized benchmarking of machine learning models. |
| KA-GNN [13] | Model Architecture | Integrates KANs into GNNs for node, edge, and graph-level processing. | Developing accurate and interpretable graph models with strong theoretical guarantees. |
| ACS Training Scheme [2] | Training Method | Adaptive checkpointing for multi-task GNNs to mitigate negative transfer. | Reliable model training in ultra-low-data regimes and with imbalanced tasks. |
The field of molecular representation is evolving toward richer, more physically grounded, and multi-faceted paradigms. The integration of geometric information, the application of novel mathematical frameworks like KANs, and the fusion of multiple data modalities are pushing the boundaries of what is possible in computational molecular science. Selecting the appropriate representation is the critical first step in building ML models that can reliably accelerate the discovery of new molecules and materials.
Molecular property prediction stands as a cornerstone of modern computational chemistry, enabling the accelerated discovery of novel pharmaceuticals, materials, and energy solutions. Machine learning (ML) models have emerged as powerful tools for predicting molecular properties, but their performance and generalizability are fundamentally constrained by the quality, breadth, and composition of the datasets on which they are trained. Understanding the landscape of available molecular property datasets—including their strengths, inherent biases, and methodological limitations—is therefore a critical prerequisite for effective research in this domain. This guide provides a comprehensive technical overview of popular molecular property datasets, systematically categorizes their inherent biases, and outlines experimental protocols to mitigate these challenges, framed within the broader context of initiating ML research for molecular property prediction.
The following table summarizes key characteristics of major molecular property datasets, highlighting their scope, common applications, and illustrative examples.
Table 1: Overview of Major Molecular Property Datasets
| Dataset Name | Primary Focus / Property Types | Approximate Size (Molecules) | Notable Features / Use Cases | Example Properties |
|---|---|---|---|---|
| OMol25 (Open Molecules 2025) [17] [12] | Quantum chemical calculations for neural network potentials | Over 100 million calculations | High-accuracy DFT (ωB97M-V/def2-TZVPD) on diverse structures; targets biomolecules, electrolytes, metal complexes [17]. | Potential energy surfaces, atomic forces, energies of large systems [17]. |
| MoleculeNet [18] [19] [2] | Curated benchmark for multiple property types | Varies by sub-dataset (e.g., 600-4200 in common sets) [18] | Aggregates multiple public sources; standardized benchmarks for ML model evaluation [19]. | Quantum mechanics, physical chemistry, biophysics (e.g., ESOL, FreeSolv, BACE) [18] [19]. |
| FGBench [19] | Functional group-level property reasoning | 625K question-answer pairs | Focuses on impact of single and multiple functional groups on properties; includes atom-level localization [19]. | Property changes from functional group additions/deletions [19]. |
| OMC25 (Open Molecular Crystals 2025) [10] | Molecular crystal structures and properties | Over 27 million structures | Dispersion-inclusive DFT relaxation trajectories of organic molecular crystals [10]. | Crystal structure properties, lattice energies [10]. |
| Therapeutic Data Commons (TDC) [20] | ADME (Absorption, Distribution, Metabolism, Excretion) and toxicity | Varies by sub-dataset | Focus on preclinical drug discovery safety and pharmacokinetic parameters [20]. | Half-life, clearance, toxicity endpoints [20]. |
Inherent Biases in Molecular Property Datasets
Despite their utility, all molecular property datasets contain inherent biases that can severely compromise model performance and generalizability if not properly addressed.
Chemical Space and Structural Biases
Datasets often suffer from limited diversity and uneven coverage of the chemical space. Early datasets were frequently restricted to simple organic molecules with only a handful of elements [17]. Although newer datasets like OMol25 have made significant strides by including biomolecules, electrolytes, and metal complexes, gaps remain in areas like polymer chemistry [12]. This structural bias means models may perform poorly on molecule classes underrepresented in training data. Furthermore, the source of molecular structures can introduce bias; for instance, datasets built primarily from commercial compound libraries may overrepresent certain structural motifs while underrepresenting natural products or novel scaffolds.
Experimental and Annotation Biases
Significant distributional misalignments often exist between different data sources for the same property, arising from variations in experimental protocols, measurement conditions, or biological assays [20]. Naive integration of such heterogeneous data can introduce noise and degrade model performance rather than improve it [20]. For ADME properties in particular, inconsistencies have been identified between gold-standard sources and commonly used benchmarks like TDC [20]. These annotation biases are especially problematic in low-data regimes where researchers must aggregate multiple sources to achieve sufficient training volume.
Property Value Distribution Biases
The distribution of property values within a dataset creates another critical bias. Most ML models excel at interpolation but struggle with extrapolation, making it difficult to identify molecular extremes—precisely the candidates often sought in materials and drug discovery [18]. When training data lacks representation of certain property value ranges (e.g., exceptionally high binding affinity or extreme stability), models cannot reliably predict these out-of-distribution (OOD) values [18]. This range limitation fundamentally constrains a model's utility in virtual screening for novel materials or drugs with exceptional characteristics.
Methodologies for Bias Assessment and Mitigation
Workflow for Dataset Evaluation and Bias Mitigation
Implementing a systematic workflow for dataset evaluation is crucial for robust molecular property prediction. The following diagram outlines key stages from initial dataset analysis to model training, highlighting steps to identify and address common biases.
Key Experimental Protocols
Data Consistency Assessment (DCA) Protocol
Purpose: To identify distributional misalignments, outliers, and annotation discrepancies across datasets before integration [20]. Tools: AssayInspector package or similar custom analysis [20]. Procedure:
- Input Preparation: Compile datasets from multiple sources for the target property. Include molecular structures (SMILES) and measured property values.
- Statistical Comparison: Calculate descriptive statistics (mean, standard deviation, quartiles) for each data source. Perform pairwise two-sample Kolmogorov-Smirnov tests to compare property value distributions across sources [20].
- Chemical Space Analysis: Generate molecular descriptors (e.g., ECFP4 fingerprints, RDKit 2D descriptors) and project into lower-dimensional space using UMAP. Visually inspect for clustering by data source rather than chemical similarity [20].
- Overlap Analysis: Identify molecules present in multiple sources and quantify annotation differences. Flag significant discrepancies for further investigation.
- Report Generation: Document all inconsistencies, including datasets with significantly different distributions, conflicting annotations, or divergent chemical spaces.
Protocol for Out-of-Distribution (OOD) Property Prediction
Purpose: To enhance model capability to extrapolate to property values outside the training distribution [18]. Method: Bilinear Transduction for zero-shot extrapolation [18]. Procedure:
- Data Partitioning: Split data into training and test sets, ensuring the test set contains property values outside the range represented in the training data.
- Model Training:
- Instead of learning to predict property values directly, train the model to learn how property values change as a function of differences in material representations [18].
- Reparameterize the prediction problem: during inference, predict property values based on a chosen training example and the representation-space difference between it and the new sample [18].
- Evaluation: Assess performance using OOD-specific metrics including extrapolative precision (fraction of true top candidates correctly identified) and recall of high-performing OOD candidates [18].
Protocol for Low-Data Regime Modeling
Purpose: To achieve accurate predictions when labeled data is extremely scarce [2]. Method: Adaptive Checkpointing with Specialization (ACS) for multi-task graph neural networks [2]. Procedure:
- Architecture Setup: Implement a shared graph neural network (GNN) backbone with task-specific multi-layer perceptron (MLP) heads.
- Training with Checkpointing:
- Train the model on all available tasks simultaneously.
- Monitor validation loss for each task independently.
- Checkpoint the best backbone-head pair for each task whenever its validation loss reaches a new minimum [2].
- Inference: Use the specialized backbone-head pair for each task during prediction.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Software Tools and Methodologies for Molecular Property Prediction
| Tool / Method Name | Type | Primary Function | Application Context |
|---|---|---|---|
| AssayInspector [20] | Software Package | Data consistency assessment; detects distributional misalignments, outliers, and batch effects across datasets. | Preprocessing before model training; identifying dataset discrepancies in ADME and physicochemical properties [20]. |
| Bilinear Transduction [18] | Machine Learning Method | Enables zero-shot extrapolation to out-of-distribution (OOD) property values. | Virtual screening for materials/molecules with property values beyond the training distribution [18]. |
| ACS (Adaptive Checkpointing with Specialization) [2] | Training Scheme | Mitigates negative transfer in multi-task learning under data imbalance. | Accurate modeling in ultra-low data regimes (e.g., <30 samples per task) [2]. |
| ChemXploreML [5] | Desktop Application | User-friendly ML for property prediction without programming; includes molecular embedders. | Rapid prototyping and prediction of physical chemical properties by experimental chemists [5]. |
| Functional Group Annotation (e.g., AccFG) [19] | Analysis Method | Precisely annotates and localizes functional groups within molecules. | Building interpretable, structure-aware models and analyzing structure-activity relationships [19]. |
The evolving landscape of molecular property datasets presents both unprecedented opportunities and significant challenges for ML researchers. While foundational datasets like MoleculeNet provide standardized benchmarks, and massive new resources like OMol25 offer unprecedented quantum chemical data, success in this field requires careful attention to the inherent biases within these datasets. Researchers must systematically address limitations in chemical space coverage, experimental inconsistencies, and property value distributions through rigorous assessment protocols and specialized methodological approaches. By implementing the data consistency checks, OOD prediction strategies, and low-data regime techniques outlined in this guide, researchers can build more robust, reliable, and generalizable molecular property prediction models that accelerate scientific discovery across chemistry, materials science, and drug development.
Understanding the Applicability Domain of a Model
In machine learning for molecular property prediction, the Applicability Domain (AD) refers to the region of chemical space where the model's predictions are expected to be reliable and accurate [21]. The concept is fundamental to ensuring trustworthy predictions in drug discovery and materials science, where decisions based on model outputs can significantly impact research outcomes and resource allocation. According to the OECD principles for model validation, defining the AD is a crucial requirement for quantitative structure-activity relationship (QSAR) models, emphasizing that reliable predictions are generally limited to chemicals structurally similar to those used in model training [22].
The core challenge stems from a fundamental machine learning limitation: models are typically trained on specific types or ranges of data, and their performance often degrades when applied to structurally dissimilar compounds [23] [24]. This is particularly problematic in molecular property prediction, where the vastness of synthesizable chemical space means that most potential compounds will be distant from previously characterized molecules [24]. Understanding and defining the AD allows researchers to identify the boundaries of their models, recognize potentially unreliable predictions, and make informed decisions about model application.
Core Methodologies for Defining Applicability Domains
Taxonomy of AD Methods
Various methodologies have been developed to define and characterize the applicability domain of predictive models. These approaches can be broadly classified into several categories based on their underlying principles and implementation strategies [22].
Table: Classification of Applicability Domain Methods
| Method Category | Core Principle | Representative Techniques | Key Advantages | Main Limitations |
|---|---|---|---|---|
| Range-Based | Defines boundaries based on descriptor ranges in training data | Bounding Box, PCA Bounding Box [22] | Simple to implement and interpret | Cannot identify empty regions or descriptor correlations |
| Geometric | Establishes geometric boundaries enclosing training data | Convex Hull [22] | Clear boundary definition | Computationally complex for high dimensions; ignores internal data distribution |
| Distance-Based | Measures similarity/distance to training set representatives | Leverage, k-NN, Mahalanobis Distance [22] [21] | Intuitive similarity measures | Threshold selection can be arbitrary; depends on distance metric choice |
| Density-Based | Estimates probability density of training data in feature space | Kernel Density Estimation (KDE) [23] | Naturally accounts for data sparsity; handles complex geometries | Bandwidth selection impacts results; computationally intensive for large datasets |
| Model-Specific | Leverages internal model characteristics for uncertainty | Bayesian Neural Networks, Ensemble Variance [21] | Directly measures prediction uncertainty | Tied to specific model architectures |
Figure 1: Taxonomy of Applicability Domain Methods
Detailed Technical Implementation
Kernel Density Estimation (KDE) Approach
Kernel Density Estimation has emerged as a powerful technique for AD determination due to its ability to naturally account for data sparsity and handle arbitrarily complex geometries of data distributions [23]. The fundamental principle involves estimating the probability density function of the training data in feature space, where regions with high density are considered in-domain, and regions with low density are considered out-of-domain.
The multivariate kernel density estimate for a query point ( x ) is given by:
[ \hat{f}h(x) = \frac{1}{n} \sum{i=1}^n Kh(x - xi) ]
where ( xi ) are the training samples, ( n ) is the number of training points, ( Kh ) is the kernel function with bandwidth ( h ). The Gaussian kernel is commonly used:
[ K_h(u) = \frac{1}{(2\pi)^{d/2}h^d} \exp\left(-\frac{\|u\|^2}{2h^2}\right) ]
where ( d ) is the dimensionality of the feature space. The bandwidth parameter ( h ) controls the smoothness of the density estimate and is typically optimized via cross-validation [23].
Distance-Based Methods
Distance-based methods are among the most widely used approaches for AD definition, particularly in QSAR modeling [22]. These methods calculate the distance of query compounds from a reference point in the training data descriptor space, with several distance metrics commonly employed:
Mahalanobis Distance: Accounts for descriptor correlations through the covariance matrix [ D_M(x) = \sqrt{(x - \mu)^T \Sigma^{-1} (x - \mu)} ] where ( \mu ) is the mean of training descriptors and ( \Sigma ) is the covariance matrix [22].
k-Nearest Neighbor (k-NN) Distance: Measures proximity to the closest training instances, with common variants including:
- ( \kappa ): Distance to the k-th nearest neighbor
- ( \gamma ): Mean distance to k-nearest neighbors
- ( \delta ): Length of the mean vector to k-nearest neighbors [21]
Leverage: Based on the hat matrix in regression analysis [ hi = xi^T (X^T X)^{-1} x_i ] where ( X ) is the model matrix of training data [21].
Bayesian and Ensemble Methods
Model-specific approaches leverage internal model characteristics to estimate prediction uncertainty. Bayesian Neural Networks provide a principled framework for uncertainty quantification by learning probability distributions over model weights rather than point estimates [21]. The predictive uncertainty can be captured through techniques such as Monte Carlo dropout or variational inference.
Similarly, ensemble methods generate uncertainty estimates by measuring the variance in predictions across multiple models [21]. The standard deviation of predictions from a heterogeneous or homogeneous ensemble serves as an indicator of model confidence for a given input.
Experimental Protocols and Validation Frameworks
Benchmarking AD Methods
Rigorous evaluation of applicability domain methods requires standardized protocols and metrics. Recent research has established comprehensive frameworks for comparing AD techniques across multiple models and datasets [21]. A typical validation workflow involves:
- Dataset Curation: Collecting diverse molecular datasets with experimentally validated properties
- Model Training: Developing QSAR models using various algorithms (random forests, neural networks, etc.)
- AD Method Application: Implementing multiple AD techniques on the trained models
- Performance Assessment: Evaluating how effectively each AD method identifies unreliable predictions
Table: Experimental Framework for AD Method Validation
| Validation Component | Implementation Details | Evaluation Metrics |
|---|---|---|
| Data Splitting | Scaffold-based splits to assess extrapolation capability | Coverage rate, domain size |
| Domain Definition | Four domain types: chemical, residual (point/group), uncertainty [23] | Precision, recall, F1-score |
| Threshold Selection | Percentile-based, error-based, or density-based cutoffs | ROC curves, precision-recall curves |
| Performance Correlation | Relationship between AD measures and prediction errors | Spearman correlation, monotonicity assessment |
| Comparative Analysis | Benchmark against multiple baseline methods | Relative improvement, statistical significance |
Case Study: CYP2B6 Inhibition Prediction
A practical implementation of AD analysis was demonstrated in a study aiming to expand the applicability domain for CYP2B6 inhibition prediction [25] [26]. The experimental protocol included:
Data Collection: CYP2B6 inhibition IC50 values were downloaded from ChEMBL and binarized (active: IC50 ≤ 10 μM; inactive: IC50 > 10 μM), resulting in 100 active and 401 inactive compounds [26].
Applicability Domain Definition: A distance-based approach defined the AD using Euclidean distance on molecular fingerprints (MACCS keys). Chemical diversity was visualized using t-distributed stochastic neighbor embedding (t-SNE) plots [25].
Domain Expansion Strategy: A drug repurposing library was screened to identify plates with the highest average minimum Euclidean distance from the training set. Selected compounds were tested experimentally for CYP2B6 inhibition [26].
Model Retraining and Evaluation: New experimental data was incorporated into the training set, and model performance was re-evaluated using one-class classification to assess domain expansion efficacy [26].
The results demonstrated that while intentional expansion of the applicability domain did not substantially increase model performance, it successfully identified new CYP2B6 inhibitors (vilanterol and allylestrenol) and increased training set diversity [25].
Protocol for KDE-Based AD Assessment
A general protocol for implementing KDE-based applicability domain assessment includes these critical steps [23]:
- Feature Selection: Choose relevant molecular descriptors (e.g., ECFP, physicochemical properties)
- KDE Training:
- Standardize features to zero mean and unit variance
- Optimize bandwidth parameter via cross-validation
- Fit KDE model to training data
- Threshold Determination:
- Calculate density values for training compounds
- Set density threshold based on desired coverage (e.g., 5th percentile of training densities)
- Domain Classification:
- For query molecules, compute density estimate using trained KDE
- Classify as in-domain if density exceeds threshold, out-of-domain otherwise
- Validation:
- Assess correlation between density values and prediction errors
- Verify that out-of-domain compounds show higher errors and chemical dissimilarity
Table: Key Research Reagents and Computational Tools
| Tool/Resource | Type | Function/Purpose | Example Applications |
|---|---|---|---|
| MACCS Keys | Molecular Descriptor | 166-bit structural keys encoding substructural features | Chemical space analysis, similarity assessment [26] |
| ECFP Fingerprints | Molecular Descriptor | Extended-Connectivity Fingerprints capturing circular substructures | Similarity searching, machine learning features [27] |
| t-SNE | Visualization Algorithm | Dimensionality reduction for chemical space visualization | Comparing training and test set distributions [25] |
| KDE Implementation | Statistical Tool | Non-parametric density estimation in high-dimensional spaces | Determining data-dense regions in feature space [23] |
| Bayesian Neural Networks | Modeling Framework | Provides uncertainty estimates alongside predictions | Confidence estimation for molecular property prediction [21] |
| One-Class Classification | Validation Method | Determines if new data belongs to training distribution | Evaluating applicability domain boundaries [26] |
| Federated Learning Platforms | Collaborative Framework | Enables multi-institutional model training without data sharing | Expanding chemical space coverage for ADMET models [28] |
Advanced Applications and Future Directions
AD for Generative Molecular Design
The concept of applicability domains is extending beyond predictive models to generative artificial intelligence for molecular design [27]. In this context, the AD defines the chemical space where generative models produce structures with acceptable drug-likeness and synthesizability. Research has explored various AD definitions for generative models, combining structural similarity to training compounds, physicochemical property similarity, unwanted substructure filters, and quantitative drug-likeness estimates (QED) [27].
Studies demonstrate that appropriate AD definitions strongly influence the drug-likeness of AI-generated molecules. Molecular Turing tests, where generated molecules are evaluated by medicinal chemists alongside human-designed compounds, provide validation of whether generative AD definitions successfully produce chemically plausible structures [27].
Federated Learning for Domain Expansion
Federated learning has emerged as a promising approach for expanding model applicability domains by enabling collaborative training across distributed datasets without centralizing sensitive data [28]. This technique is particularly valuable for ADMET prediction, where individual organizations typically have limited data coverage of relevant chemical space.
Key benefits of federated learning for AD expansion include:
- Altering the geometry of chemical space a model can learn from, improving coverage
- Systematic outperformance of local baselines, with improvements scaling with participant diversity
- Expanded applicability domains, with increased robustness across unseen scaffolds
- Persistent benefits across heterogeneous data sources and assay protocols [28]
Large-scale initiatives like the MELLODDY project have demonstrated that cross-pharma federated learning at unprecedented scale unlocks benefits in QSAR without compromising proprietary information [28].
Figure 2: Applicability Domain Assessment Workflow
Defining and understanding the applicability domain of molecular property prediction models is not merely an academic exercise but a practical necessity for reliable drug discovery and materials science research. As machine learning continues to transform molecular design, robust AD methods provide the guardrails that enable responsible application of these powerful technologies. The continuing evolution of AD methodologies—from simple distance-based approaches to sophisticated density estimation and Bayesian uncertainty quantification—reflects the growing recognition that knowing when not to trust a model is as important as knowing when to trust it. For researchers beginning their journey in molecular property prediction, establishing rigorous practices for applicability domain assessment should be considered an essential component of any modeling workflow.
Building Your Model: Predictive and Generative Modeling Approaches
The accurate prediction of molecular properties is a critical challenge in accelerating drug discovery and materials science. Traditional experimental methods are often associated with significant costs and time investments [5]. Machine learning (ML) has emerged as a powerful tool to mitigate these burdens; however, selecting the appropriate modeling approach is paramount for success. This guide provides a structured framework for researchers and drug development professionals to choose between two fundamental ML paradigms: deterministic and probabilistic modeling. The core distinction lies in their treatment of uncertainty. Deterministic models provide single, point estimates for a given molecule, while probabilistic models output a distribution of possible values, thereby quantifying the model's confidence in its own predictions [29] [30]. Within the context of molecular property prediction—a field often characterized by small, sparse, and noisy datasets—understanding and leveraging this distinction is key to building reliable and informative models [31] [32].
Core Conceptual Differences
At its heart, the choice between a deterministic and probabilistic model is a choice about how to handle uncertainty. This section breaks down the fundamental characteristics of each approach.
Deterministic Machine Learning
Deterministic models operate on predefined rules and logic. Given the same input (a molecular representation), a deterministic system will always produce the same output (a predicted property value) [33]. These models are trained to optimize a scalar-valued loss function, such as mean squared error or cross-entropy, and provide a single best estimate for each input [29].
Key Characteristics:
- Predictability: Outputs are entirely determined by the inputs and the programmed model weights.
- Transparency: The logic behind the system's decision-making is often more easily understandable and auditable.
- Limited Adaptability: Cannot inherently express confidence or learn from uncertainty without structural changes [30] [33].
Probabilistic Machine Learning
Probabilistic models, in contrast, incorporate uncertainty and express outcomes as likelihoods rather than certainties. They use statistical models to analyze data and provide a probabilistic characterization of their predictions [29] [34]. Instead of a single value, the output may be a distribution (e.g., a mean and variance for a continuous property) which allows the model to know what it does not know [29].
Key Characteristics:
- Statistical Reasoning: Uses probability theory to express confidence in outcomes.
- Adaptability: Can evolve as new data becomes available and is designed to work with ambiguous or incomplete information.
- Uncertainty Quantification (UQ): Provides a natural framework for quantifying predictive uncertainty, which is crucial for identifying unreliable predictions [30] [32].
The table below summarizes the core differences between these two approaches.
Table 1: Core Differences Between Deterministic and Probabilistic Models
| Factor | Deterministic Model | Probabilistic Model |
|---|---|---|
| Output Type | Single, point estimate (e.g., a property value) | Probability distribution (e.g., mean and variance) |
| Uncertainty Handling | Does not quantify its own uncertainty | Explicitly quantifies predictive uncertainty |
| Data Requirements | Requires complete, clean data to function optimally | Tolerates incomplete or noisy data better |
| Transparency | Easy to audit and explain due to fixed logic paths | Can be a "black box"; may require tools for explainability |
| Primary Strength | Precision and predictability in well-defined scenarios | Pattern recognition and decision-making under uncertainty |
Molecular Property Prediction Context
The theoretical differences between deterministic and probabilistic models have profound practical implications in molecular property prediction. Drug discovery pipelines are long and complex, with a low overall success rate, creating a strong business need for technologies that can lower attrition and costs [35]. ML models are applied across all stages, from identifying novel targets and optimizing small-molecule compounds to analyzing digital pathology data [35].
A significant challenge in this domain is that the effectiveness of ML is often limited by scarce and incomplete experimental datasets [31]. This "low-data regime" makes it difficult for models to generalize well. Furthermore, poor predictive accuracy is often related to two key issues:
- Regions of chemical space with steep structure-activity relationships (SAR), where small structural changes lead to large property differences.
- A lack of representation of test molecules in the training data [32].
Probabilistic models directly address these challenges by providing a measure of reliability for their predictions. This allows researchers to flag molecules where the model's prediction is likely to be inaccurate, either because the molecule is too different from the training set or because it lies in a structurally complex region of the chemical space [32]. As noted in recent research, "reliable methods to quantify the predictive uncertainty of machine learning models can significantly increase the impact of molecular property prediction" in applications like active learning and ML-guided property optimization [32].
Technical Comparison and Selection Framework
Choosing the right model is a contextual decision. The following table and workflow diagram provide a structured guide for researchers.
Table 2: Model Selection Guide for Molecular Property Prediction
| Decision Factor | Deterministic Model | Probabilistic Model |
|---|---|---|
| Data Quality & Availability | Large, high-quality datasets with precise labels and consistent identifiers. | Smaller, noisier, or fragmented datasets; data with inherent uncertainty. |
| Regulatory & Audit Needs | High-priority; decisions must be explainable and reproducible (e.g., for regulatory filings). | Less critical, or can be addressed with additional explainability tools (e.g., SHAP values). |
| Task Nature | Prediction of properties where a single, best answer is required and uncertainty is low. | Prediction in complex SAR regions or for applications like active learning, where understanding confidence is key. |
| Desired Output | A single property value for each molecule (e.g., predicted IC50). | A value with a confidence interval (e.g., IC50 ± SD) or a full probability distribution. |
| Example Applications | Initial high-throughput screening where speed and a clear cutoff are needed. | Prioritizing compounds for synthesis in a lead optimization campaign, where knowing the confidence can prevent wasted resources. |
Diagram 1: Model Selection Workflow
Experimental Protocols and Implementations
To ground the theoretical comparison, this section outlines detailed methodologies for implementing both model types in a molecular property prediction task.
A Deterministic CNN for Property Prediction
A deterministic approach can be implemented using a Convolutional Neural Network (CNN) that operates on molecular graph representations or other structured data [29] [35].
Architecture:
- Input Layer: Accepts a featurized representation of the molecule (e.g., a graph, fingerprint, or image).
- Convolutional Layer: Performs a convolutional operation with a set of filters and a ReLU activation function to extract local molecular features.
- Max-Pooling Layer: Reduces the spatial dimensionality of the feature maps, retaining the most salient information.
- Flatten Layer: Converts the pooled feature maps into a one-dimensional vector.
- Dense (Output) Layer: A fully-connected layer with a linear activation function (for regression) or softmax (for classification) to produce the final, single-value prediction [29].
Training Protocol:
- Loss Function: Mean Squared Error (MSE) for regression tasks; Sparse Categorical Crossentropy for classification.
- Optimizer: RMSprop or Adam.
- Training: The model is trained to minimize the difference between its scalar output and the experimental value for each molecule in the training set [29].
A Probabilistic Model with Uncertainty Quantification
A probabilistic model extends the deterministic architecture to output a distribution. One effective method is to model the output as a Gaussian distribution.
Architecture:
- Feature Extraction Layers: Identical to the deterministic CNN (Convolutional, Max-Pooling, Flatten).
- Dense Layer: A fully-connected layer that maps the flattened features to a higher-dimensional space.
- Probabilistic Output Layer: This layer is configured to output two parameters: the mean (μ) and the variance (σ²) of a Gaussian distribution. The loss function is then the negative log-likelihood of the training data under this predicted distribution [29] [34] [32].
Training Protocol:
- Loss Function: Negative Log-Likelihood (NLL). This function penalizes the model both for incorrect mean predictions and for high variance when it should be confident (and vice versa).
- Optimizer: RMSprop or Adam.
- Training: The model learns to predict a distribution for each molecule. The mean represents the most likely property value, and the variance represents the model's uncertainty in that prediction [29] [32].
Diagram 2: Deterministic vs. Probabilistic Model Architectures
Successfully implementing ML models for molecular property prediction requires a suite of computational tools and datasets.
Table 3: Essential Research Reagents for Molecular Property Prediction
| Tool / Resource | Type | Function in Research |
|---|---|---|
| TensorFlow with TensorFlow Probability | Programmatic Framework | Provides a flexible, open-source ecosystem for building and training both deterministic and probabilistic deep learning models [29] [35]. |
| PyTorch | Programmatic Framework | An alternative open-source ML library popular for research, offering dynamic computation graphs and robust support for deep learning [35]. |
| ChemXploreML | Desktop Application | A user-friendly, offline-capable app that allows chemists to make property predictions without deep programming expertise, automating molecular featurization [5]. |
| Graph Neural Networks (GNNs) | Model Architecture | A specialized neural network architecture that operates directly on graph-based molecular structures, often achieving state-of-the-art results [31] [36]. |
| Multi-task Learning | Training Methodology | A technique to improve model generalization by training a single model on multiple, related property prediction tasks simultaneously, which is especially useful with sparse data [31]. |
| QM9 Dataset | Benchmark Dataset | A public dataset containing quantum-mechanical properties for ~134,000 small organic molecules, commonly used for training and benchmarking models [31]. |
| Active Learning Loop | Experimental Design | A process that uses a model's uncertainty estimates (from a probabilistic model) to intelligently select which compounds to test next in an experiment, maximizing information gain [32]. |
The journey to selecting the right modeling approach for molecular property prediction is a strategic one. Deterministic models offer simplicity, speed, and transparency, making them suitable for well-defined problems with abundant, high-quality data. However, the inherent challenges of molecular data—its sparsity, noise, and complex structure-activity landscapes—often make probabilistic models the more robust and informative choice. By quantifying predictive uncertainty, probabilistic models empower researchers to make risk-aware decisions, prioritize experimental resources effectively, and ultimately increase the impact of machine learning in accelerating drug discovery and materials design. A modern research workflow may even leverage both, using deterministic models for initial screening and probabilistic models for finer, more critical optimization tasks. The key is to align the model's capabilities with the project's specific data context, regulatory requirements, and strategic goals.
Model Architectures for Different Representations
Molecular property prediction stands as a cornerstone in accelerated drug discovery and materials science. The paradigm has shifted from traditional descriptor-based machine learning to geometric deep learning, where models directly learn from molecular graph structures. This transition enables more accurate capture of intricate topological and chemical information, moving beyond the limitations of manual feature engineering [37]. The core challenge lies in selecting and implementing model architectures that align with specific molecular representations and property characteristics. This guide provides a comprehensive technical overview of contemporary architectures, their experimental protocols, and performance benchmarks to inform researchers' model selection and development.
Foundational Architectures and Their Representations
Core Architectural Paradigms
Different molecular properties stem from distinct structural and geometric characteristics, necessitating specialized model architectures for their prediction. The following table summarizes the primary architectural families, their core principles, and ideal application domains.
Table 1: Foundational Model Architectures for Molecular Property Prediction
| Architecture | Core Principle | Molecular Representation | Ideal for Property Types |
|---|---|---|---|
| Graph Isomorphism Network (GIN) [37] | Uses injective aggregation functions to capture local node neighborhoods and substructures, providing high expressiveness for graph isomorphism. | 2D topological graph (atoms=nodes, bonds=edges). | Properties determined by molecular topology/functional groups (e.g., carcinogenicity in MUTAG [37]). |
| Equivariant GNN (EGNN) [37] | Incorporates 3D atomic coordinates and preserves Euclidean symmetries (translation, rotation, reflection) in its operations. | 3D geometric graph (includes spatial atom coordinates). | Quantum chemical and spatially-sensitive properties (e.g., dipole moment, Air-Water Partition Coefficient - log Kaw [37]). |
| Graphormer [37] | Integrates graph topology with global attention mechanisms, allowing nodes to interact directly based on structural encoding. | 2D/3D graph enhanced with spatial encoding. | Complex properties requiring long-range dependency modeling (e.g., bioactivity on OGB-MolHIV [37]). |
| Kolmogorov-Arnold GNN (KA-GNN) [13] | Replaces standard MLPs with learnable, interpretable univariate functions (e.g., Fourier series) on graph edges within the GNN pipeline. | Standard 2D molecular graph. | General molecular prediction with enhanced interpretability and parameter efficiency [13]. |
| Multi-Task GNN with ACS [2] | Combines a shared GNN backbone with task-specific heads and adaptive checkpointing to mitigate negative transfer in multi-task learning. | 2D molecular graph. | Predicting multiple properties simultaneously, especially in ultra-low data regimes for specific tasks [2]. |
Architectural Selection Workflow
The following diagram outlines the logical decision process for selecting an appropriate model architecture based on the property of interest and available data.
Diagram 1: Model Selection Workflow
Advanced and Specialized Frameworks
Kolmogorov-Arnold Networks (KANs) for Graphs
KA-GNNs represent a recent innovation by integrating KAN modules into the three fundamental components of GNNs: node embedding, message passing, and readout. Unlike traditional multi-layer perceptrons (MLPs) that use fixed activation functions on nodes, KANs place learnable univariate functions (e.g., Fourier series, B-splines) on edges [13]. This design offers superior expressivity, parameter efficiency, and inherent interpretability. The Fourier-series-based functions in KA-GNNs are particularly effective for capturing both low-frequency and high-frequency structural patterns in molecular graphs, providing strong theoretical approximation guarantees grounded in Fourier analysis and Carleson's theorem [13].
Experimental Protocol for KA-GNNs:
- Architecture Variants: Two primary variants are developed: KA-Graph Convolutional Networks (KA-GCN) and KA-Graph Attention Networks (KA-GAT). In KA-GCN, initial node embeddings are created by passing concatenated atomic and neighboring bond features through a KAN layer. Message passing follows the GCN scheme, with node updates via residual KANs. KA-GAT further incorporates edge embeddings initialized with KAN layers [13].
- Training & Evaluation: Models are evaluated across seven diverse molecular benchmarks. Performance is measured by prediction accuracy (e.g., ROC-AUC, MAE) and computational efficiency. Interpretability is qualitatively assessed by the model's ability to highlight chemically meaningful substructures [13].
- Key Results: KA-GNNs consistently outperform conventional GNNs in accuracy and computational efficiency. The learned network mappings often align with known chemical principles, providing valuable insights for lead optimization [13].
Frameworks for Imperfect and Multi-Task Data
Real-world molecular datasets are often imperfectly annotated, meaning most properties are labeled for only a subset of molecules. The OmniMol framework addresses this by modeling the entire molecule-property universe as a hypergraph, where each property is a hyperedge connecting all molecules annotated with it [38]. This structure captures three key relationships: molecule-molecule, molecule-property, and property-property.
Experimental Protocol for OmniMol:
- Architecture: Built upon a Graphormer backbone, OmniMol integrates a task-routed Mixture of Experts (t-MoE) that uses task embeddings to dynamically activate specialized model pathways. This enables task-adaptive predictions with O(1) complexity, independent of the number of tasks. It also includes an SE(3)-equivariant encoder to incorporate 3D molecular conformation and enforce physical symmetries [38].
- Training: The model is trained end-to-end on all available molecule-property pairs. A scale-invariant message passing strategy and equilibrium conformation supervision are used to facilitate learning-based conformational relaxation [38].
- Key Results: OmniMol achieves state-of-the-art performance on 47 out of 52 ADMET-P (Absorption, Distribution, Metabolism, Excretion, Toxicity - Physicochemical) prediction tasks and demonstrates strong chirality awareness, which is critical for drug safety [38].
For multi-task learning, the Adaptive Checkpointing with Specialization (ACS) scheme effectively mitigates negative transfer. Negative transfer occurs when updates from one task degrade performance on another, a common issue in multi-task learning with imbalanced data [2].
Experimental Protocol for ACS:
- Architecture: A single, shared GNN backbone learns general-purpose molecular representations. These feed into separate, task-specific MLP heads.
- Training Scheme: Unlike standard MTL that saves one final model, ACS continuously monitors the validation loss for each task. It checkpoints the best backbone-head pair for a task whenever that task's validation loss hits a new minimum. This results in a specialized model for each task, protecting them from detrimental parameter updates later in training [2].
- Key Results: On benchmarks like ClinTox, SIDER, and Tox21, ACS matched or surpassed state-of-the-art supervised methods. It demonstrated particular efficacy in ultra-low data regimes, accurately predicting sustainable aviation fuel properties with as few as 29 labeled samples per task [2].
Benchmarking and Performance
Quantitative benchmarking is essential for evaluating model efficacy across diverse property types. The following table synthesizes key performance metrics from comparative analyses of major architectures.
Table 2: Architectural Benchmarking on Standardized Tasks
| Architecture | Dataset & Task | Key Metric | Reported Performance | Comparative Context |
|---|---|---|---|---|
| Graphormer [37] | OGB-MolHIV (Bioactivity) | ROC-AUC | 0.807 | Best performance on this bioactivity classification task. |
| Graphormer [37] | MoleculeNet (log Kow) | Mean Absolute Error (MAE) | 0.18 | Best performance for this partition coefficient. |
| EGNN [37] | MoleculeNet (log Kaw) | MAE | 0.25 | Best performance for this geometry-sensitive property. |
| EGNN [37] | MoleculeNet (log K_d) | MAE | 0.22 | Best performance for this soil-water partition coefficient. |
| GIN [37] | MUTAG (Carcinogenicity) | Accuracy | ~0.90 (inferred) | Not explicitly stated, but performs well on topology-based tasks. |
| ACS Scheme [2] | ClinTox (FDA approval/Toxicity) | Average Improvement | +15.3% vs. STL | Effective mitigation of negative transfer in multi-task setting. |
| KA-GNN [13] | Multiple Benchmarks (7 datasets) | Prediction Accuracy | Consistent outperformance vs. GNNs | Superior accuracy and computational efficiency. |
The Scientist's Toolkit
Implementing and evaluating these models requires a standardized set of software tools, datasets, and molecular featurization methods.
Table 3: Essential Research Reagents for Molecular Property Prediction
| Tool / Resource | Type | Primary Function | Example in Context |
|---|---|---|---|
| MoleculeNet [19] [37] | Benchmark Dataset Collection | Provides standardized datasets for fair model comparison across quantum mechanics, physical chemistry, and biophysics. | Used as the primary benchmark for models like GIN, EGNN, and Graphormer [37]. |
| OGB (Open Graph Benchmark) [37] | Benchmark Dataset Collection | Provides large-scale, realistic benchmark datasets for graph ML. | OGB-MolHIV is a key dataset for evaluating bioactivity prediction [37]. |
| Functional Group Annotations | Molecular Featurization | Provides fine-grained, chemically meaningful substructures that link structure to property. | The FGBench dataset provides 625K problems with FG annotations to enhance LLM and GNN reasoning [19]. |
| Graph Kernel Algorithms | Similarity Metric | Quantifies structural similarity between molecules from a global perspective. | Used in MSSM-GNN to build a similarity graph, enhancing molecular representation learning [39]. |
| Adaptive Checkpointing (ACS) [2] | Training Scheme | Mitigates negative transfer in multi-task learning with imbalanced data. | Enabled accurate prediction of fuel properties with only 29 labeled samples [2]. |
| SE(3)-Equivariant Encoder [38] | Model Component | Encodes 3D molecular conformation while respecting physical symmetries (rotation/translation). | A core component of OmniMol, enabling chirality-aware predictions without expert features [38]. |
The field of molecular property prediction has evolved beyond one-size-fits-all models towards specialized architectures tailored to the nature of the target property and data landscape. As evidenced, selecting the right model depends critically on whether the property is rooted in 2D topology, 3D geometry, or requires global attention. Furthermore, practical challenges like imperfect annotation and multi-task interference are now being addressed by innovative frameworks like OmniMol and ACS. Mastering this diverse toolkit of architectures and their respective strengths is fundamental for researchers aiming to deploy machine learning effectively in drug discovery and materials science. Future progress will likely involve a deeper fusion of these architectural paradigms and a stronger emphasis on data-efficient, explainable models that can reliably guide experimental efforts.
The fundamental goal of de novo molecular design is to identify novel compounds with desired properties from an virtually infinite chemical space, estimated to contain between 10^60 and 10^100 potential drug-like molecules [40]. This search task is computationally intractable using traditional methods, necessitating sophisticated computational approaches. Recent advances in artificial intelligence (AI) and machine learning (ML) have revolutionized this field, enabling researchers to generate novel molecular structures efficiently and predict their properties with increasing accuracy [41]. This technical guide explores the core paradigms, methodologies, and practical implementations of generative models for molecular design, framed within the broader context of initiating ML research for molecular property prediction—a critical prerequisite for effective generative design.
The integration of generative models into the drug discovery pipeline represents a paradigm shift from traditional screening-based approaches to automated design. However, the efficacy of these models relies heavily on accurate molecular property prediction, which faces significant challenges including data scarcity, imbalanced datasets, and the need for robust validation frameworks [2]. Understanding these challenges and the solutions being developed, such as multi-task learning and specialized architectures, provides the essential foundation for successful implementation of de novo molecular design systems.
Fundamentals of Molecular Representation
Before implementing generative models, molecules must be translated into numerical representations comprehensible to machine learning algorithms. This translation process is a critical first step in both property prediction and generative design.
Molecular Representations and Descriptors
- SMILES Strings: Simplified Molecular-Input Line-Entry System (SMILES) provides a linear notation representing molecular structure using ASCII strings, enabling the application of natural language processing techniques to chemical structures [40].
- Molecular Fingerprints: Binary vectors that encode the presence or absence of specific molecular substructures or patterns. RDKit provides multiple fingerprint types including Morgan fingerprints (equivalent to ECFP), RDKit fingerprints, and MACCS keys [42].
- Molecular Graph Representations: Graph-based representations treat atoms as nodes and bonds as edges, preserving the topological structure of molecules and enabling the application of graph neural networks (GNNs) [2].
- Molecular Descriptors: Computational chemistry features such as molecular weight, logP, topological polar surface area, and Lipinski rule counts, which can be calculated using toolkits like RDKit and used as input for predictive models [42].
Comparison of Molecular Representations
Table 1: Comparison of Molecular Representation Approaches for Machine Learning
| Representation Type | Format | Advantages | Limitations | Common Use Cases |
|---|---|---|---|---|
| SMILES Strings | Text/Sequence | Simple, compact, widely supported | May represent same molecule differently; validity challenges | Transformer-based generation, sequence models |
| Molecular Fingerprints | Fixed-length binary vectors | Fast similarity search, well-established | May miss structural nuances, fixed dimensionality | Similarity screening, QSAR models, classification |
| Molecular Graphs | Graph (nodes+edges) | Preserves topological structure, natural representation | Computationally intensive, complex model architectures | Graph Neural Networks, property prediction |
| 3D Coordinate Sets | Atomic coordinates & elements | Encodes spatial conformation, essential for some properties | Conformational flexibility, alignment sensitivity | Structure-based design, docking studies |
Core Paradigms in Generative Molecular Design
De novo molecular design strategies can be categorized according to the coarseness of their molecular representation, each with distinct advantages and implementation considerations [41].
Atom-Based Generation
Atom-based approaches generate molecules atom by atom, providing maximum flexibility but requiring careful constraint management to ensure chemical validity.
- Implementation: Typically uses recurrent neural networks (RNNs) or transformers that sequentially add atoms to a growing molecular structure, with rules to maintain chemical validity during the generation process.
- Advantages: Maximum exploration of chemical space, ability to discover truly novel scaffolds not predefined by fragments.
- Challenges: High computational complexity, potential for generating invalid structures without careful constraint implementation, slower generation times.
Fragment-Based Generation
Fragment-based methods assemble molecules from predefined chemical fragments or building blocks, enhancing the likelihood of generating synthetically accessible and drug-like molecules.
- Implementation: Uses fragment libraries derived from known drug molecules, with generative models learning to combine these fragments in novel ways while maintaining chemical validity.
- Advantages: Higher probability of synthetic accessibility, more drug-like molecules, faster generation times compared to atom-based approaches.
- Challenges: Limited by the diversity of the fragment library, potential bias toward known chemical space.
Reaction-Based Generation
Reaction-based approaches employ knowledge of chemical reactions to assemble molecules, prioritizing synthetic feasibility by mimicking how chemists actually construct molecules in the laboratory.
- Implementation: Incorporates reaction rules and synthetic pathways into the generation process, often using retrosynthetic analysis principles to design molecules that can be efficiently synthesized.
- Advantages: Highest synthetic accessibility, direct integration with medicinal chemistry practices, reduced synthesis failure rates.
- Challenges: Requires comprehensive reaction databases, complex implementation, may limit novelty due to reliance on known reaction templates.
Comparative Analysis of Design Paradigms
Table 2: Performance Comparison of Molecular Design Paradigms
| Design Paradigm | Chemical Validity Rate | Novelty | Synthetic Accessibility | Representative Model Types |
|---|---|---|---|---|
| Atom-Based | 50-90% (with constraints) | High | Variable | RNN, Transformer, GAN |
| Fragment-Based | 85-100% | Moderate | High | GAN, VAE, Reinforcement Learning |
| Reaction-Based | 95-100% | Moderate | Very High | Template-based, Transformer |
Practical Implementation and Workflows
Transformer-Based Generative Architecture
Recent advances have adapted transformer architectures, originally developed for natural language processing, for molecular generation. These models treat SMILES strings as sequences and learn to generate novel, valid molecular structures [40].
Transformer-based Molecular Generation
Multi-Task Learning for Property Prediction
Accurate property prediction is essential for evaluating generated molecules. Multi-task learning (MTL) addresses data scarcity by leveraging correlations among related molecular properties, but faces challenges with negative transfer in imbalanced datasets [2].
Adaptive Checkpointing with Specialization Workflow
Experimental Protocols and Methodologies
Implementing Adaptive Checkpointing with Specialization (ACS)
The ACS training scheme mitigates negative transfer in multi-task learning while preserving benefits of parameter sharing [2]:
Architecture Setup: Implement a shared graph neural network (GNN) backbone with task-specific multi-layer perceptron (MLP) heads. The GNN learns general-purpose molecular representations while MLP heads provide task-specific learning capacity.
Training Procedure: Train the model using a combined loss function with masking for missing labels. Monitor validation loss for each task independently throughout the training process.
Checkpointing Mechanism: Save model parameters whenever any task achieves a new minimum validation loss. This creates specialized backbone-head pairs for each task, protecting against detrimental parameter updates from other tasks.
Validation Protocol: Use scaffold-based splitting (e.g., Murcko scaffolds) to ensure structurally dissimilar training and test sets, providing more realistic performance estimates compared to random splits.
Evaluation Metrics: Assess performance using task-specific metrics (ROC-AUC, precision-recall, RMSE) and compare against single-task learning and conventional MTL baselines.
Transformer-Based Molecular Generation Protocol
For implementing transformer-based de novo molecular design [40]:
Data Preparation: Curate a dataset of validated SMILES strings representing drug-like molecules. Apply standardization and canonicalization to ensure consistent representation.
Model Architecture: Implement a transformer encoder-decoder architecture with multi-head attention mechanisms. For target-specific generation, condition the attention mechanism on target protein representations.
Training Procedure: Train using teacher forcing with cross-entropy loss on SMILES tokens. Incorporate techniques such as scheduled sampling to improve generation robustness.
Conditional Generation: For target-specific generation, implement different keys and values in the multi-head attention for each target protein, allowing the generation process to be conditioned on specific biological targets.
Validation and Filtering: Implement chemical validity checks using toolkit like RDKit, and assess generated molecules for drug-likeness, novelty, and target-specific properties.
Cheminformatics Platforms and Toolkits
Table 3: Essential Research Tools for Molecular Property Prediction and Design
| Tool/Platform | Type | Key Features | Molecular Representation Support | Licensing |
|---|---|---|---|---|
| RDKit | Open-source Cheminformatics Library | Molecular fingerprints, descriptor calculation, substructure search, scaffold analysis | SMILES, Molecular graphs, Fingerprints | BSD-3 Open Source |
| ChemAxon Suite | Commercial Cheminformatics Platform | Chemical database management, ADMET prediction, reactivity prediction | Multiple representations | Commercial |
| ChemXploreML | Desktop ML Application | Graphical interface for property prediction, molecular embedders, offline capability | Molecular descriptors, Embeddings | Freeware |
| AutoDock Vina | Molecular Docking Software | Protein-ligand docking, binding affinity prediction | 3D molecular structures | Apache Open Source |
Benchmark Datasets for Method Validation
- MoleculeNet Benchmarks: Standardized datasets including ClinTox (FDA approval vs. toxicity), SIDER (side effects), and Tox21 (toxicity endpoints) for fair comparison of molecular property prediction methods [2].
- Sustainable Aviation Fuel (SAF) Properties: Real-world dataset with 15 physicochemical properties demonstrating capability in ultra-low data regimes (as few as 29 labeled samples) [2].
- ADMET Property Databases: Curated datasets for absorption, distribution, metabolism, excretion, and toxicity properties essential for drug discovery.
Future Directions and Challenges
While generative models for molecular design have shown remarkable progress, several challenges remain to be addressed:
- Data Scarcity and Quality: Ultra-low data regimes continue to pose challenges for property prediction, particularly for novel molecular classes with limited experimental data [2].
- Multi-Objective Optimization: Simultaneous optimization of multiple, often competing, molecular properties remains computationally challenging and requires advanced Pareto-front exploration techniques.
- Synthetic Accessibility Prediction: Improved assessment and guidance of synthetic feasibility during the generation process to ensure practical utility of designed molecules.
- Interpretability and Explainability: Developing methods to provide chemical insights into why specific molecular structures are generated and how they relate to target properties.
- Integration with Experimental Workflows: Creating seamless pipelines between computational design and experimental validation to accelerate iterative design-make-test-analyze cycles.
The field continues to evolve rapidly, with emerging opportunities in target-specific compound generation [40], few-shot learning for low-data scenarios, and integration of generative design with automated synthesis and testing platforms. By addressing these challenges and leveraging the methodologies outlined in this guide, researchers can accelerate the discovery of novel molecular structures with tailored properties for pharmaceutical, materials, and energy applications.
Advanced Feature Selection with Differentiable Information Imbalance (DII)
Feature selection is a critical preprocessing step in machine learning for molecular property prediction, essential for preventing overfitting, increasing model performance, and obtaining interpretable models. The high dimensionality of molecular data, often comprising thousands of features from genomic sequences, molecular descriptors, or quantum chemical calculations, presents significant analytical challenges. This is particularly acute in drug discovery, where accurately predicting molecular behavior from limited samples is paramount [43] [44].
Conventional feature selection methods—wrapper, embedded, and filter techniques—often struggle with key uncertainties: determining the optimal number of features for a simplified yet informative model, and aligning features with different units and relative importance [45]. These challenges are pronounced in molecular sciences, where data points may combine heterogeneous features like atomic distances (nanometers) and hydrogen bond counts (dimensionless) [45].
Differentiable Information Imbalance (DII) represents a major advance in addressing these limitations. This automated method ranks feature set information content and learns optimal feature weights through gradient descent, simultaneously performing unit alignment and importance scaling. By preserving essential information in a lower-dimensional space, DII facilitates the creation of simplified, interpretable models crucial for scientific discovery [45] [46].
Core Concepts: From Information Imbalance to DII
The Information Imbalance Framework
The foundation of DII is the Information Imbalance Δ, a measure quantifying the predictive power between two distance metrics [45]. Formally, for a dataset with two feature space representations (A) and (B), the Information Imbalance (\Delta(d^A \to d^B)) measures how well distances in space (A) predict distances in space (B):
[ \Delta(d^A \to d^B) = \frac{2}{N^2} \sum{i,j: r{ij}^A = 1} r_{ij}^B ]
Here, (r{ij}^A) and (r{ij}^B) represent the distance ranks of data point (j) from (i) according to distance metrics (d^A) and (d^B) respectively [45]. The sum is taken over all pairs where (j) is the nearest neighbor of (i) in space (A).
A DII value close to 0 indicates that space (A) is an excellent predictor of space (B), as nearest neighbors in (A) remain nearest neighbors in (B). Conversely, a value near 1 suggests space (A) provides no information about space (B), with ranks distributed uniformly [45].
Differentiable Information Imbalance (DII)
The standard Information Imbalance is a powerful comparative tool, but its non-differentiable nature regarding feature weights limits optimization. DII overcomes this by creating a differentiable loss function, enabling direct gradient-based optimization of feature weights [45].
Given a ground truth feature space (B) and a parameterized feature space (A(\mathbf{w})) with weights (\mathbf{w}), DII minimizes (\Delta(d^{A(\mathbf{w})} \to d^B)) through gradient descent. This allows the algorithm to automatically learn:
- Optimal feature weights accounting for different units and intrinsic importance
- Sparse solutions through L1 regularization, effectively performing feature selection
- The optimal size of the reduced feature space [45]
Table 1: Key Advantages of DII Over Traditional Feature Selection Methods
| Aspect | Traditional Methods | DII Approach |
|---|---|---|
| Unit Alignment | Often requires manual preprocessing or normalization | Automatically learns scaling weights to align different units |
| Importance Weighting | Typically relies on statistical heuristics | Optimizes importance weights directly via gradient descent |
| Optimal Feature Count | Usually requires cross-validation or manual specification | Can determine optimal size automatically through sparsity constraints |
| Interpretability | Varies by method; often black-box | Preserves interpretability through linear feature weighting |
Implementation and Workflow
The DII Optimization Algorithm
The DII optimization process follows these key steps [45]:
- Initialization: Begin with initial feature weights, typically set to unity
- Distance Calculation: Compute weighted distances between all data points in the parameterized feature space
- Rank Determination: Calculate distance ranks for all data point pairs
- Gradient Computation: Compute the gradient of the DII loss function with respect to the feature weights
- Weight Update: Adjust weights via gradient descent to minimize the DII
- Convergence Check: Repeat steps 2-5 until convergence or a maximum number of iterations
The algorithm can operate in both supervised and unsupervised modes. When a separate ground truth feature space is provided, it functions as a supervised method. When the full input feature set serves as the ground truth, it operates unsupervised [45].
Practical Implementation
DII is publicly available in the DADApy Python library [45] [47]. The implementation includes comprehensive documentation and tutorials for applying DII to various molecular systems [45].
The following diagram illustrates the complete DII workflow for molecular property prediction:
DII Molecular Workflow: The complete DII optimization pipeline for molecular property prediction, from feature extraction to final model building.
Experimental Protocols and Validation
Benchmarking DII on Molecular Systems
The effectiveness of DII has been demonstrated through two primary molecular applications [45]:
Identifying Collective Variables (CVs)
Objective: Identify optimal low-dimensional descriptors of biomolecular conformations from high-dimensional feature spaces [45].
Methodology:
- Input Features: Molecular dynamics trajectories described by numerous structural parameters (dihedral angles, interatomic distances, etc.)
- Ground Truth: Full feature space or expert-curated CVs
- Optimization: DII with sparsity constraints to select minimal feature sets
- Validation: Compare free energy landscapes and kinetic properties against ground truth
Feature Selection for Machine Learning Force Fields
Objective: Select optimal subsets of Atom-Centered Symmetry Functions (ACSFs) for training Behler-Parrinello neural network potentials for liquid water [45].
Methodology:
- Input Features: Extensive library of ACSF descriptors
- Ground Truth: Smooth Overlap of Atomic Position (SOAP) descriptors, known as high-quality representations
- Optimization: DII to identify most informative ACSF subsets
- Validation: Force field accuracy and computational efficiency compared to using full ACSF sets
Comparative Performance Analysis
DII has demonstrated superior performance against traditional feature selection methods in molecular applications. The table below summarizes key quantitative comparisons:
Table 2: Performance Comparison of Feature Selection Methods in Molecular Applications
| Method | Application Context | Key Advantages | Limitations |
|---|---|---|---|
| DII | Collective variable identification; Force field feature selection | Automated unit alignment; Optimal feature weighting; Sparse solutions | Requires differentiable distance metric; Gradient-based optimization |
| Knowledge-Based | Drug response prediction using pathway genes [44] | High interpretability; Biological relevance | Limited to prior knowledge; May miss novel features |
| PCA | Dimensionality reduction for drug response [44] | Captures maximum variance; Computationally efficient | Linear assumptions; Limited interpretability |
| Transcription Factor Activities | Drug response prediction on tumor data [44] | Biological relevance; Effective for certain drug classes | Limited to transcriptional regulation |
| Autoencoders | Nonlinear feature transformation [44] | Captures complex patterns; Flexible architecture | Computational intensity; Potential overfitting |
The Scientist's Toolkit: Essential Research Reagents
Implementing DII for molecular property prediction requires several key computational tools and resources:
Table 3: Essential Research Tools for DII Implementation
| Tool/Resource | Function | Implementation Notes |
|---|---|---|
| DADApy Library | Python implementation of DII [45] [47] | Primary software package; includes tutorials and documentation |
| Molecular Features | Input features for DII optimization | Can include distances, angles, chemical descriptors, or omics measurements |
| Ground Truth Space | Reference space for DII optimization | High-fidelity descriptors (e.g., SOAP) or full feature set [45] |
| Distance Metric | Basis for Information Imbalance calculation | Euclidean distance typically used; must be differentiable |
| Optimization Framework | Gradient descent implementation | Standard deep learning frameworks (PyTorch, TensorFlow) compatible |
| Validation Metrics | Performance assessment | Prediction accuracy, model interpretability, computational efficiency |
Integration with Molecular Property Prediction Workflows
Integrating DII into molecular property prediction pipelines addresses critical bottlenecks in computational drug discovery. The method is particularly valuable in scenarios with:
High-Dimensional Multi-Omics Data: DII can identify predictive biomarker panels from thousands of genomic, transcriptomic, and proteomic features, as demonstrated in drug response prediction challenges [43].
Limited Training Data: By selecting maximally informative features, DII improves model generalization in data-scarce regimes common to molecular property prediction [2].
Interpretability Requirements: The clear feature weighting and selection provided by DII offers biological insights beyond black-box predictions, crucial for understanding drug mechanisms [44].
Recent advances demonstrate DII's compatibility with state-of-the-art molecular representation learning approaches, including graph neural networks and message-passing architectures [4]. This positions DII as a versatile component in modern molecular machine learning pipelines, from initial feature selection to final predictive modeling.
Navigating Pitfalls: Data, Uncertainty, and Model Robustness
Addressing the Small Data Regime and Dataset Bias
Molecular property prediction is a cornerstone of modern drug discovery and materials science, enabling researchers to screen vast chemical spaces in silico. However, the efficacy of machine learning (ML) in this domain is often constrained by two interconnected challenges: the small data regime and dataset bias. The small data problem arises because acquiring high-quality, experimental property data for molecules is often costly, time-consuming, and technically challenging [48] [49]. This data scarcity is frequently compounded by dataset bias, where training data is not representative of the broader chemical space or real-world application scenarios due to historical research focus, experimental convenience, or systemic biases in data collection [50] [51].
For researchers beginning ML research in molecular property prediction, understanding and addressing these challenges is not optional—it is fundamental to building reliable, robust, and equitable models. This guide provides a technical foundation for navigating the small data regime while systematically identifying and mitigating bias, ensuring your research is both impactful and ethically sound.
Understanding the Small Data Challenge
In the context of molecular property prediction, "small data" refers to datasets with a limited number of labeled molecular compounds, which is often the case for many physicochemical, biological, or toxicological properties [2] [48]. The acquisition of molecular data is constrained by factors such as the high cost of experiments, ethical considerations, privacy, security, and technical limitations [48].
The primary consequence of small data is the sharp decrease in the predictive performance of ML models, primarily due to overfitting, where a model learns the noise in the training data rather than the underlying pattern [48]. Table 1 summarizes the core problems and their implications for model development.
Table 1: Core Challenges in the Small Data Regime and Their Implications
| Challenge | Description | Impact on Model Performance |
|---|---|---|
| Data Scarcity | Insufficient labeled samples for the target property [48]. | High variance in predictions, leading to poor generalization to new, unseen molecules. |
| Data Imbalance | Severe task imbalance where certain properties have far fewer labels than others in multi-task settings [2]. | Degraded efficacy of multi-task learning through negative transfer; models become biased towards properties with more data. |
| High Dimensionality | The number of molecular descriptors or features is large compared to the number of samples [49]. | Increased risk of overfitting; the "curse of dimensionality" makes it difficult to learn meaningful patterns. |
| Data Diversity | The available data covers only a limited region of the chemical space of interest [48]. | Models fail to make accurate predictions for molecules with scaffolds or functional groups not represented in the training set. |
Technical Strategies for Small Data
A range of ML strategies has been developed to overcome the limitations of small data. These can be broadly categorized into data-centric approaches, algorithmic solutions, and novel learning strategies.
Data-Centric Approaches
These methods focus on augmenting or making better use of existing data.
Multi-task Learning (MTL): MTL leverages correlations among related molecular properties to improve predictive performance by training a single model on multiple tasks simultaneously [2] [31]. Through inductive transfer, MTL allows the model to use information from one task to benefit another. However, imbalanced datasets can lead to negative transfer, where updates from one task degrade performance on another [2]. The Adaptive Checkpointing with Specialization (ACS) training scheme has been developed to mitigate this. ACS combines a shared, task-agnostic graph neural network backbone with task-specific heads, adaptively saving the best model parameters for each task when its validation loss is minimized [2]. This approach has been validated on molecular property benchmarks, showing an 11.5% average improvement over other node-centric message passing methods and enabling accurate predictions with as few as 29 labeled samples [2].
Transfer Learning and Pre-trained Models: This involves leveraging knowledge from a data-rich source task (e.g., a large, general molecular dataset) to improve learning on a data-scarce target task. While pre-trained models offer strong performance, they require significant computational resources for pre-training and may need fine-tuning for domain-specific targets [2].
Data Augmentation: This includes generating new training examples through techniques like Generative Adversarial Networks (GANs) or physical model-based augmentation, which uses simulations to create synthetic data [48].
Table 2: Summary of Small Data Mitigation Strategies
| Strategy | Mechanism | Best-Suited For |
|---|---|---|
| Multi-task Learning (MTL) | Shares representations and leverages signals across correlated prediction tasks [2] [31]. | Projects with multiple, potentially related, molecular properties to predict. |
| Transfer Learning | Transfers knowledge from a model trained on a large, general dataset to a specific, low-data task [2] [48]. | Scenarios where a large, relevant source dataset is available for pre-training. |
| Active Learning | Iteratively selects the most informative data points for labeling from a pool of unlabeled samples, optimizing the learning process [52] [49]. | Settings where acquiring new data is possible but expensive, and the goal is to minimize labeling cost. |
| Semi-Supervised & Self-Supervised Learning | Leverages unlabeled data to learn better representations, which can then be used for the primary supervised task with limited labels [48]. | Situations with abundant unlabeled molecular data but scarce labeled data for the target property. |
Algorithmic and Modeling Solutions
Choosing or designing the right model is critical for small data.
Model-Agnostic Sampling: This technique identifies an optimal training data distribution that leads to the highest accuracy for a model of a given, constrained size. It challenges the conventional wisdom that training and test data must come from the same distribution, especially for small, interpretable models [53].
Uncertainty Quantification and Active Learning: For ML-driven molecular dynamics, uncertainty-biased MD can be used to enhance data collection. This approach biases simulations toward regions where the model's predictions are uncertain, simultaneously capturing rare events and extrapolative regions to build more comprehensive training sets [52]. Conformal Prediction (CP) is a valuable technique for calibrating these uncertainties, ensuring they are accurately aligned with prediction errors and preventing the exploration of unphysical configurations [52].
The following diagram illustrates the workflow of an active learning cycle that integrates these concepts for building robust interatomic potentials.
Identifying and Mitigating Dataset Bias
Bias in molecular data can lead to models that are inaccurate, unreliable, and perpetuate existing inequalities in healthcare and materials science [50]. Bias can be defined as any systematic and unfair difference in how predictions are generated for different populations, leading to disparate outcomes [50] [51].
Types and Origins of Bias
Bias can enter the AI lifecycle at multiple stages, from human origins to deployment.
Human Biases:
- Implicit Bias: Subconscious attitudes or stereotypes that become embedded in how data is collected or labeled [50]. For example, clinical trial data may over-represent certain demographic groups.
- Systemic Bias: Broader institutional norms or policies that lead to inequities, such as inadequate funding for research on diseases affecting minority populations [50].
- Confirmation Bias: The tendency of model developers to seek or interpret data in a way that confirms their pre-existing beliefs or hypotheses [50].
Algorithm Development Biases:
- Representation Bias: Occurs when the training data over-represents certain molecular scaffolds or compound classes and under-represents others, often reflecting historical research interests [50] [51].
- Temporal Bias & Training-Serving Skew: Arises when the relationship between molecular structure and property changes between the time of training data collection and model deployment (concept shift), or when the data distributions differ [50].
A systematic framework is essential for identifying and mitigating these biases. The following workflow, inspired by the Biological Bias Assessment Guide [51], provides a structured approach.
Mitigation Strategies
During Data Collection and Preparation:
- Documentation: Use Datasheets for Datasets to document the motivation, composition, and collection process of your molecular datasets. This promotes transparency and helps researchers understand potential biases [51].
- Diverse Data Sourcing: Actively seek to include diverse molecular structures and, when relevant, diverse demographic information to combat representation bias [50].
During Model Development and Evaluation:
- Stratified Evaluation: Move beyond aggregate performance metrics. Evaluate model performance separately across different molecular scaffolds, functional groups, and, for biomedical applications, demographic subgroups [50] [51].
- Fairness Metrics: Incorporate metrics like demographic parity, equalized odds, and counterfactual fairness to quantitatively assess and enforce fairness in predictions [50].
- Adherence to Guidelines: Follow consensus-based checklists like the REFORMS guidelines to improve the transparency, reproducibility, and validity of ML-based science [51].
Experimental Protocols and the Scientist's Toolkit
This section provides a practical roadmap for implementing the strategies discussed.
Protocol: Implementing Multi-task Learning with ACS
This protocol is adapted from studies that successfully applied MTL to molecular property prediction in low-data regimes [2] [31].
Problem Formulation & Data Preparation:
- Define the primary target property (the small-data task) and identify potential auxiliary tasks.
- Data Source: Collect data from molecular databases (e.g., ChEMBL, PubChem) or high-throughput experiments [49].
- Featurization: Represent molecules as graphs. Nodes are atoms (with features like element type, hybridization), and edges are bonds (with features like bond type) [2].
- Splitting: Split the dataset using a Murcko-scaffold split to ensure that molecules with different core structures are separated between training, validation, and test sets. This provides a more realistic estimate of real-world performance [2].
Model Architecture Setup:
Training with Adaptive Checkpointing:
- Use a masked loss function to handle missing property labels for some molecules.
- Monitor the validation loss for each task independently throughout the training process.
- For each task, save a checkpoint of the entire model (shared backbone + its specific head) whenever its validation loss hits a new minimum.
- This process yields a specialized model for each task, mitigating negative transfer [2].
Evaluation:
- Report the primary metric (e.g., ROC-AUC, RMSE) for the target property on the held-out test set.
- Compare the ACS model against strong baselines, including a Single-Task Learning model and a standard MTL model without checkpointing [2].
The Scientist's Toolkit
Table 3: Essential Resources for Molecular Property Prediction Research
| Tool / Resource | Function | Relevance to Small Data/Bias |
|---|---|---|
| Graph Neural Networks (GNNs) | Deep learning architecture that operates directly on molecular graph structures [2]. | The backbone for modern MTL approaches like ACS, enabling effective knowledge transfer between tasks. |
| High-Throughput Computation/Experimentation | Automated, parallelized methods for generating large amounts of molecular data [49]. | Addresses data scarcity at the source by systematically expanding the available data. |
| Uncertainty Quantification Methods | Techniques like ensembles or gradient-based features to estimate the reliability of a model's prediction [52]. | Core to active learning; helps identify data points for labeling and regions where the model is likely to fail. |
| Model Cards & Data Cards | Documentation frameworks for transparently reporting model and dataset characteristics [51]. | Key bias mitigation tools; force critical consideration of intended use cases, limitations, and data composition. |
| Active Learning Cycles | Iterative protocol that combines model training, uncertainty-based sampling, and targeted data acquisition [52]. | A strategic framework for minimizing experimental costs while maximizing model performance and coverage of chemical space. |
| Domain Knowledge Descriptors | Feature engineering based on expert knowledge (e.g., physiochemical properties) [49]. | Can improve model interpretability and performance when data is limited, by providing strong inductive biases. |
Navigating the small data regime and confronting dataset bias are defining challenges in molecular property prediction. As this guide has outlined, a successful research approach is multifaceted. It involves selecting sophisticated learning strategies like MTL with ACS and active learning, rigorously evaluating models for biased performance across chemical and demographic spaces, and adhering to frameworks that promote transparency and reproducibility.
By integrating these technical strategies and ethical considerations from the outset, researchers can build models that are not only accurate on average but are also robust, reliable, and equitable. This foundational work is critical for accelerating the discovery of new therapeutics and materials in a responsible and effective manner.
The Critical Impact of Dataset Size on Representation Learning Models
In molecular property prediction, the scarcity of high-quality, labeled data is a fundamental constraint that shapes the selection, design, and performance of machine learning models. This guide examines the critical relationship between dataset scale and model efficacy, detailing how modern representation learning techniques—including self-supervised learning (SSL), multi-task learning (MTL), and foundation models—are engineered to overcome data limitations. By providing structured protocols and resource toolkits, this document equips researchers with practical strategies to navigate the low-data regimes prevalent in drug discovery, enabling more robust and predictive modeling of molecular properties.
The pursuit of artificial intelligence-driven drug discovery is inherently linked to the availability of standardized, high-quality biological and chemical data. Biology's complexity necessitates data that captures changes across cells, tissues, and organs in response to disease or medication; however, public datasets have often fallen short in reliably training AI models [54]. The efficacy of molecular property prediction models is fundamentally constrained by the availability and quality of training data [2]. In many practical domains, including pharmaceutical drugs and green energy carriers, the scarcity of reliable labels impedes the development of robust predictors [2]. This data bottleneck is exacerbated in preclinical safety modeling, where limited data and experimental heterogeneity create significant integration challenges that can compromise predictive accuracy [20]. Understanding how representation learning techniques leverage both labeled and unlabeled data is therefore paramount for advancing molecular machine learning.
Quantitative Landscape: Dataset Scale and Model Performance
The relationship between dataset size, model architecture, and task performance follows predictable yet complex patterns. The following table summarizes key quantitative findings from recent research on dataset scaling effects:
Table 1: Impact of Dataset Scale on Model Performance and Strategies
| Model / Technique | Dataset Size / Characteristics | Performance Impact / Key Finding | Citation |
|---|---|---|---|
| Self-Supervised Representation Learning (SSRL) for EHR data | Review of 46 studies (2019-2024); Model families: Transformers (43%), Autoencoders (28%), GNNs (17%) | Reduces need for manual labeling; enables generalization across tasks without full retraining | [55] |
| ACS (Adaptive Checkpointing with Specialization) for MTL | Accurate predictions with as few as 29 labeled samples (Sustainable Aviation Fuel properties) | Mitigates negative transfer in imbalanced training datasets; outperforms single-task learning and conventional MTL in low-data regimes | [2] |
| Traditional Machine Learning (ML) | Effective with hundreds to thousands of labeled examples | Performs well on small-to-medium structured datasets; superior for tabular data in cost and accuracy | [56] |
| Deep Learning (DL) | Requires large-scale labeled datasets (often millions) for effective generalization | Essential for unstructured data (text, images, audio); provides better representations for complex tasks | [56] |
| Data Integration (e.g., AqSolDB solubility) | Integration nearly doubled molecular coverage | Improved model performance through expanded chemical space and increased samples | [20] |
Analysis of public ADME datasets reveals significant distributional misalignments and annotation discrepancies between gold-standard and popular benchmark sources. Naive integration of these heterogeneous datasets often degrades model performance despite increasing sample size, highlighting that data consistency is as critical as data volume [20].
Core Technical Mechanisms: Learning from Limited Labels
Self-Supervised Representation Learning (SSRL)
SSRL automatically discovers and extracts features from unlabeled data by training algorithms to predict part of the data from other parts. The model learns to 'recover' whole or partial information from its original input, enabling it to identify patterns and structures within unlabeled data to produce efficient representation vectors [55]. In healthcare applications, SSRL has demonstrated particular value for Electronic Health Records (EHR) categorical data, which is easier to de-identify and enables faster construction of large datasets while maintaining patient privacy [55]. The learned representations serve as inputs for subsequent predictive models, reducing the need for extensive manual labeling and often outperforming models trained on similar volumes of labeled data [55].
Multi-Task Learning with Adaptive Checkpointing (ACS)
MTL leverages correlations among related molecular properties to alleviate data bottlenecks through inductive transfer. However, its effectiveness is often undermined by negative transfer (NT)—performance drops that occur when updates driven by one task are detrimental to another [2]. The recently introduced Adaptive Checkpointing with Specialization (ACS) approach integrates a shared, task-agnostic backbone with task-specific trainable heads, adaptively checkpointing model parameters when NT signals are detected [2]. During training, the backbone is shared across tasks, and after training, a specialized model is obtained for each task [2]. This design promotes inductive transfer among sufficiently correlated tasks while protecting individual tasks from deleterious parameter updates, enabling accurate predictions with as few as 29 labeled samples in real-world scenarios like sustainable aviation fuel property prediction [2].
Foundation Models and Representation Potentials
Foundation models, trained through large-scale pretraining on vast and heterogeneous data, have driven remarkable progress by acquiring highly transferable and general-purpose representations [57]. A growing body of research indicates that the representations learned by foundation models exhibit strong similarity across architectures, training objectives, and even modalities—a capacity termed "representation potential" [57]. This characteristic enables them to serve as versatile starting points for downstream molecular prediction tasks, significantly reducing the required labeled data for specific applications. Foundation models are distinguished by three key features: (1) training on broad, web-scale datasets; (2) use of self-supervision to learn directly from raw, unlabeled data; and (3) adaptability to wide-ranging downstream tasks through fine-tuning or prompting [57].
Experimental Protocols for Low-Data Regimes
Protocol: ACS for Molecular Property Prediction
Objective: To implement Adaptive Checkpointing with Specialization for predicting multiple molecular properties with limited labeled data.
Materials:
- Molecular dataset with multiple property annotations (e.g., ClinTox, SIDER, Tox21)
- Graph Neural Network architecture
- Task-specific multi-layer perceptron heads
Methodology:
- Data Preparation:
Model Architecture:
- Implement a shared GNN backbone based on message passing [2]
- Attach dedicated task-specific MLP heads for each molecular property
- Initialize with shared parameters across all tasks
Training Procedure:
- Monitor validation loss for every task independently
- Checkpoint the best backbone-head pair whenever a task's validation loss reaches a new minimum
- Employ early stopping based on aggregated performance metrics
Evaluation:
- Compare against baselines including single-task learning (STL), MTL without checkpointing, and MTL with global loss checkpointing (MTL-GLC)
- Assess performance gains particularly for tasks with the fewest labeled samples
Validation: ACS has demonstrated an average 11.5% improvement relative to other node-centric message passing methods and outperforms STL by 8.3% on average, with particularly large gains (15.3%) on the ClinTox dataset [2].
Protocol: Data Consistency Assessment with AssayInspector
Objective: To systematically evaluate dataset compatibility before integration for molecular property prediction.
Materials:
- Multiple molecular property datasets (e.g., half-life data from Obach et al., Lombardo et al., Fan et al.)
- AssayInspector package (publicly available at https://github.com/chemotargets/assay_inspector)
Methodology:
- Data Collection:
- Gather molecular property data from diverse sources (e.g., TDC, ChEMBL, literature-curated sets)
- Standardize molecular representations and property annotations
Statistical Analysis:
- Generate descriptive statistics for each dataset (mean, standard deviation, quartiles)
- Perform two-sample Kolmogorov-Smirnov tests for regression tasks
- Conduct Chi-square tests for classification tasks
- Compute within- and between-source feature similarity values
Visualization:
- Create property distribution plots across datasets
- Generate UMAP projections to assess chemical space coverage
- Visualize molecular overlaps and annotation discrepancies
Insight Report Generation:
- Identify dissimilar datasets based on descriptor profiles
- Flag conflicting annotations for shared molecules
- Detect divergent datasets with low molecular overlap
Validation: Studies have revealed significant misalignments between benchmark and gold-standard ADME sources, with naive integration often degrading model performance despite increased sample size [20].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Computational Tools for Molecular Representation Learning
| Tool / Resource | Type / Category | Primary Function in Research |
|---|---|---|
| ACS Training Scheme | Algorithm | Mitigates negative transfer in multi-task learning; enables learning with ultra-low data (e.g., 29 samples) [2] |
| AssayInspector | Software Package | Systematically identifies distributional misalignments and annotation discrepancies across datasets prior to modeling [20] |
| SSRL Models | Model Architecture | Learns transferable representations from unlabeled data; reduces dependency on manual labeling for molecular tasks [55] |
| Graph Neural Networks (GNNs) | Model Architecture | Operates on graph-structured molecular data; captures relational information between atoms and bonds [56] |
| Therapeutic Data Commons (TDC) | Data Resource | Provides standardized benchmarks for molecular property prediction; aggregates ADME and physicochemical data [20] |
| RxRx3-core | Dataset | Public cellular screening dataset with 222,601 microscopy images; enables benchmarking of microscopy vision models [54] |
| Foundation Models | Pre-trained Models | Provides general-purpose molecular representations through large-scale pretraining; adaptable to specific tasks with minimal fine-tuning [57] |
The critical impact of dataset size on representation learning models in molecular property prediction is undeniable, yet innovative methodologies are progressively overcoming historical data limitations. Through self-supervised learning, multi-task learning with adaptive checkpointing, and the leveraging of foundation models, researchers can extract meaningful patterns from limited labeled examples by strategically utilizing both labeled and unlabeled data. The future landscape will likely be shaped by more sophisticated data consistency assessment tools, standardized benchmarking approaches, and the continued development of representation learning techniques that explicitly address the challenges of data heterogeneity and scarcity. As these methodologies mature, they promise to accelerate the pace of AI-driven molecular discovery and design, ultimately reducing the time and cost associated with traditional drug development pipelines.
Quantifying and Managing Uncertainty in Predictions
In machine learning-based molecular property prediction, uncertainty quantification (UQ) is a critical toolkit component that enables researchers to assess the reliability of model outputs. Predictive inaccuracy often stems from two primary sources: regions of chemical space characterized by large property differences for structurally similar molecules, and insufficient representation of test molecules in the training data [32]. For drug development professionals, distinguishing between high and low-confidence predictions is essential for prioritizing compounds for synthesis and testing, particularly when exploring novel chemical spaces with limited high-quality data [59]. This technical guide provides a comprehensive framework for implementing robust uncertainty quantification methods within molecular property prediction workflows, addressing both theoretical foundations and practical applications relevant to research scientists in pharmaceutical and materials development.
Theoretical Foundations of Predictive Uncertainty
Categorizing Uncertainty Types
In molecular property prediction, uncertainties are broadly classified into two fundamental categories, each with distinct characteristics and mitigation strategies:
Aleatoric uncertainty represents the inherent noise in the data itself, arising from experimental measurement errors, variations in experimental conditions, or the intrinsic stochasticity of molecular systems [59]. This uncertainty is irreducible without improving data quality or collection methods. In chemistry applications, aleatoric uncertainty often exhibits heteroscedastic characteristics, meaning the noise level varies across different regions of chemical space, particularly when integrating data from multiple sources with varying accuracy levels [59].
Epistemic uncertainty stems from limitations in the model's knowledge, resulting from insufficient training data in certain chemical domains, inadequate model complexity, or fundamental gaps between training and application domains [59] [60]. Unlike aleatoric uncertainty, epistemic uncertainty is reducible through targeted expansion of training datasets, particularly in underrepresented regions of chemical space, or through model architecture improvements [59].
The distinction between these uncertainty types is not merely theoretical. In practice, separately quantifying aleatoric and epistemic uncertainties enables researchers to diagnose the root causes of prediction unreliability and implement appropriate mitigation strategies [59]. For instance, high epistemic uncertainty suggests the need for additional data collection in specific chemical domains, while high aleatoric uncertainty indicates fundamental limitations in data quality that may require improved experimental protocols.
Uncertainty Visualization for Scientific Interpretation
Effective communication of uncertainty information requires careful consideration of visualization strategies. Value-suppressing uncertainty palettes, which "grey out" high-uncertainty regions while maintaining color contrast for high-certainty predictions, have emerged as particularly effective for conveying uncertainty in molecular property visualizations [61]. These palettes acknowledge that when uncertainty is very high, the mean prediction value provides limited additional value to decision-makers, and thus should be visually de-emphasized.
For scientific applications, discrete color palettes with few bins that encode both value and uncertainty information on the same scale have demonstrated superior performance in user comprehension studies compared to continuous color schemes [61]. When implementing uncertainty visualization, it is crucial to ensure sufficient color contrast and to test palettes for accessibility to users with color vision deficiencies, which affects approximately 1 in 12 men and 1 in 200 women [62].
Uncertainty Quantification Methodologies
Comparative Analysis of UQ Methods
Table 1: Comparison of Uncertainty Quantification Methods for Molecular Property Prediction
| Method | Uncertainty Types Captured | Key Principles | Implementation Complexity | Computational Cost | Best-Suited Applications |
|---|---|---|---|---|---|
| Deep Ensembles [59] [63] | Both aleatoric and epistemic | Multiple models with different initializations; variance in predictions indicates uncertainty | Moderate | High (proportional to ensemble size) | Active learning, model reliability assessment, out-of-domain detection |
| Monte Carlo Dropout [59] | Primarily epistemic | Multiple stochastic forward passes with dropout enabled during inference | Low | Moderate | Scenarios requiring uncertainty with single model |
| Evidential Approaches [63] | Both (theoretically) | Places prior distributions over model parameters and updates via evidence | High | Low to Moderate | Applications requiring principled Bayesian framework |
| Quantile Regression [60] | Aleatoric | Models conditional quantiles of output distribution using asymmetric loss functions | Moderate | Low | Capturing data noise heterogeneity, confidence intervals |
| Conformal Prediction [64] | Both through coverage guarantees | Provides distribution-free confidence sets with marginal coverage guarantees | Moderate | Low to Moderate | Guaranteed confidence intervals, risk assessment |
Technical Implementation Protocols
Deep Ensembles Implementation
Deep Ensembles have emerged as a particularly robust approach for uncertainty quantification in molecular property prediction [59] [63]. The implementation protocol consists of the following key steps:
Ensemble Generation: Train multiple neural networks (typically 5-10) independently with different random initializations. Each network should be modified to output both a mean (μ(x)) and variance (σ²(x)) parameterizing a Gaussian distribution [59].
Loss Function Specification: For each network, optimize the weights by minimizing the negative log-likelihood (NLL) loss function: [ \text{NLL} = \sum{k=1}^{N} \frac{1}{2} \ln(\sigma^2(xk)) + \frac{(yk - \mu(xk))^2}{2\sigma^2(x_k)} ] This enables the network to learn both the prediction and its associated uncertainty [59].
Uncertainty Decomposition: For a query molecule, calculate the total predictive uncertainty as: [ \sigma{total}^2 = \underbrace{\frac{1}{M} \sum{m=1}^{M} \sigmam^2(x)}{\text{Aleatoric}} + \underbrace{\frac{1}{M} \sum{m=1}^{M} (\mum(x) - \bar{\mu}(x))^2}_{\text{Epistemic}} ] where M is the number of ensemble members, μₘ(x) and σₘ²(x) are the predictions from the m-th model, and μ̄(x) is the ensemble mean prediction [59].
Recent research indicates that while Deep Ensembles produce sharp uncertainty estimates, they often require post-hoc calibration to correct for systematic miscalibration, which can be addressed using techniques such as isotonic regression or standard scaling [63].
Readout Ensembling for Foundation Models
For large foundation models where full ensembling is computationally prohibitive, readout ensembling provides an efficient alternative:
Foundation Model Initialization: Start with a pre-trained foundation model (e.g., MACE-MP-0 for neural network potentials) [60].
Readout Layer Diversification: Fine-tune only the final readout layers of multiple model instances on different subsets of the target training data, maintaining the core representation layers fixed [60].
Uncertainty Calculation: Compute epistemic uncertainty as the standard deviation across the readout ensemble predictions, which captures model uncertainty while leveraging the generalized representations learned by the foundation model [60].
This approach significantly reduces computational costs compared to full ensembling while maintaining the ability to identify out-of-domain structures and quantify epistemic uncertainty [60].
Advanced UQ Applications and Extensions
Explainable Uncertainty Attribution
Beyond quantifying overall uncertainty, recent advances enable attribution of uncertainty to specific atoms within a molecule, providing chemical insights into prediction reliability:
Atom-Based Uncertainty Modeling: Adapt Deep Ensembles to output atom-level uncertainty contributions alongside molecular property predictions, enabling identification of specific chemical substructures contributing to prediction uncertainty [59].
Uncertainty Rationalization: Analyze atomic uncertainty values to diagnose chemical components introducing uncertainty, such as unrecognized functional groups or rare structural motifs underrepresented in training data [59].
Active Learning Integration: Utilize atom-attributed uncertainties to prioritize not only which compounds to test experimentally, but also which specific chemical substructures require additional data collection [59].
This explainable uncertainty framework transforms UQ from a black-box reliability metric into a chemically interpretable tool for hypothesis generation and experimental design.
Uncertainty Calibration Protocols
Raw uncertainty estimates from ML models often exhibit systematic miscalibration, where the predicted confidence intervals do not match empirical error rates. Post-hoc calibration is essential for transforming descriptive uncertainty metrics into actionable signals:
Calibration Data Preparation: Reserve a separate calibration dataset not used during model training, with representative coverage of the target chemical space [63].
Calibration Method Selection:
- Isotonic Regression: Non-parametric approach that learns a monotonic transformation of uncertainty scores to improve calibration [63].
- Standard Scaling: Learns a simple scaling factor to align predicted variances with observed errors [63].
- GP-Normal Correction: Applies Gaussian process-based normalization to uncertainty estimates [63].
Calibration Assessment: Evaluate calibration quality using reliability diagrams and proper scoring rules (e.g., negative log-likelihood, Brier score) on held-out test data [63].
Computational experiments on benchmark datasets including QM9 demonstrate that properly calibrated uncertainty estimates can improve high-confidence prediction filtering efficacy and reduce redundant ab initio evaluations in active learning by more than 20% [63].
Table 2: Uncertainty Calibration Methods and Their Applications
| Calibration Method | Key Principles | Data Requirements | Computational Complexity | Optimal Use Cases |
|---|---|---|---|---|
| Isotonic Regression [63] | Learns non-parametric monotonic transformation of scores | Moderate (1K-10K samples) | Moderate | General-purpose calibration for diverse chemical spaces |
| Standard Scaling [63] | Applies linear scaling to variance estimates | Low (100-1K samples) | Low | Scenarios with approximately normal residual distributions |
| GP-Normal Correction [63] | Gaussian process-based uncertainty normalization | Moderate to High | High | Small-molecule datasets with smooth property landscapes |
| Platt Scaling | Logistic regression-based probability calibration | Low | Low | Binary classification tasks |
Experimental Design and Data Considerations
Data Consistency Assessment
Uncertainty quantification reliability is fundamentally dependent on data quality and consistency. The AssayInspector toolkit provides a systematic approach for data consistency assessment prior to modeling:
Distributional Analysis: Apply statistical tests (e.g., two-sample Kolmogorov-Smirnov test for regression tasks) to identify significant distributional differences between data sources [20].
Chemical Space Evaluation: Use dimensionality reduction techniques (UMAP) to visualize dataset coverage and identify potential applicability domain gaps [20].
Annotation Consistency Checking: Detect conflicting property annotations for shared compounds across different datasets, which may indicate experimental protocol differences or data quality issues [20].
Analysis of public ADME datasets has revealed substantial distributional misalignments and annotation discrepancies between benchmark and gold-standard sources, highlighting that naive data integration without consistency assessment often degrades model performance despite increasing training set size [20].
Evaluation Metrics and Validation Protocols
Robust evaluation of uncertainty quantification performance requires multiple complementary metrics:
Calibration Metrics:
- Expected Calibration Error (ECE): Measures the difference between predicted confidence and empirical accuracy across confidence bins.
- Negative Log-Likelihood (NLL): Proper scoring rule that evaluates both sharpness and calibration of predictive distributions.
Sharpness Metrics:
- Mean Prediction Interval Width: Assesses the average size of confidence intervals, with narrower intervals indicating sharper predictions when calibration is maintained.
Downstream Task Performance:
- Active Learning Efficiency: Measures the reduction in experimental samples required to achieve target performance when using uncertainty-guided selection.
- Counterfactual Truthfulness: Evaluates whether uncertainty estimates improve the reliability of explanatory insights, particularly for counterfactual explanations in interpretable AI [65].
Validation should be performed under different data splitting scenarios, including random splits, scaffold splits (to test generalization to novel chemotypes), and temporal splits (to simulate real-world deployment conditions), as UQ method performance varies significantly across these evaluation settings [32].
Research Reagent Solutions
Table 3: Essential Computational Tools for Uncertainty Quantification in Molecular Property Prediction
| Tool/Category | Representative Examples | Primary Function | Application Context |
|---|---|---|---|
| Uncertainty Quantification Libraries | Deep Ensembles [59], Monte Carlo Dropout [59], Evidential Deep Learning [63] | Implement core UQ algorithms for neural networks | General molecular property prediction, active learning |
| Chemical Foundation Models | MACE-MP-0 [60], CHGNet [60], ANI-1 [60] | Provide pre-trained representations for broad chemical spaces | Transfer learning, fine-tuning for specific properties |
| Data Consistency Assessment | AssayInspector [20] | Identify dataset discrepancies and distribution misalignments | Data aggregation, quality control, federated learning |
| Calibration Tools | Isotonic Regression, Standard Scaling, GP-Normal [63] | Correct systematic miscalibration in uncertainty estimates | Post-processing of model outputs for reliable confidence intervals |
| Visualization Platforms | Viz Palette [62], Value-Suppressing Uncertainty Palettes [61] | Create accessible uncertainty visualizations | Scientific communication, decision support systems |
| Benchmark Datasets | QM9 [64] [63], ESOL, FreeSolv, Lipophilicity [64] | Provide standardized evaluation benchmarks | Method development, comparative performance assessment |
Effective uncertainty quantification and management represents a critical capability for reliable molecular property prediction in research and drug development. The methodologies outlined in this technical guide—from robust ensemble methods and explainable uncertainty attribution to systematic calibration protocols—provide a comprehensive framework for implementing trustworthy prediction systems. As molecular machine learning continues to advance, several emerging trends warrant particular attention: the development of foundation-model-specific UQ approaches that leverage pre-trained representations while quantifying epistemic uncertainty [60], improved integration of uncertainty quantification with explainable AI to enhance scientific interpretability [65], and standardized benchmarking protocols that evaluate UQ methods under realistic deployment conditions including significant distribution shifts [32] [20]. By adopting these systematic approaches to uncertainty quantification, researchers and drug development professionals can significantly enhance the reliability and actionable insights derived from molecular property prediction models.
Mitigating the Effect of Activity Cliffs on Model Performance
Molecular property prediction is a cornerstone of computer-aided drug design, enabling researchers to evaluate the potential biological activity and pharmacokinetic properties of chemical compounds before undertaking costly synthetic efforts and experimental assays [66]. At the heart of this endeavor lie Quantitative Structure-Activity Relationship (QSAR) models, which aim to establish mathematical relationships between molecular structures and their biological effects. However, the fundamental principle underlying QSAR—that structurally similar molecules exhibit similar activities—is frequently violated by a phenomenon known as activity cliffs (ACs) [67].
Activity cliffs are defined as pairs or groups of structurally similar compounds that share the same target but display large, unexpected differences in potency [67] [68]. These cliffs represent the pinnacle of structure-activity relationship (SAR) discontinuity, where minute chemical modifications lead to dramatic changes in biological activity [69]. For medicinal chemists, ACs provide rich SAR information that reveals critical chemical transformations with substantial biological impact. For machine learning models, however, ACs present significant challenges as they create discontinuities in the chemical space that are difficult to capture accurately [66].
The presence of ACs in training data is particularly problematic for Graph Neural Networks (GNNs) and other advanced deep learning approaches. When structurally similar molecules with different activities are embedded closely in latent space, models often yield poor predictions for these challenging cases [66]. Recent systematic investigations have confirmed that QSAR models frequently fail to predict ACs, and this limitation forms a major source of prediction error [69]. This whitepaper provides a comprehensive technical guide to understanding activity cliffs and implementing state-of-the-art mitigation strategies to enhance the robustness and predictive power of molecular property prediction models.
Understanding the Fundamental Challenges Posed by Activity Cliffs
Defining and Characterizing Activity Cliffs
The activity cliff concept, first mentioned by Lajiness in 1991, has evolved considerably over the past three decades [67]. While the core definition remains consistent—structurally similar compounds with large potency differences—the operationalization of this concept requires careful consideration of both similarity criteria and potency difference thresholds:
Similarity Criteria: Molecular similarity can be assessed through various computational approaches, with Tanimoto similarity based on molecular fingerprints representing the earliest and most common method [67]. More chemically intuitive approaches include Matched Molecular Pairs (MMPs), defined as pairs of compounds distinguished only by a chemical modification at a single site [67] [68]. For MMP-based ACs (termed "MMP-cliffs"), the cliff is defined when the potency difference exceeds a predefined threshold (typically a 100-fold difference, or ΔpKi ≥ 2.0) [68].
Beyond Structural Similarity: The AC concept has expanded to include three-dimensional similarity ("3D-cliffs") based on experimental ligand-protein complex structures, and interaction cliffs that capture similarities and differences in ligand-target interaction patterns [67].
Coordinated AC Formation: Rather than occurring as isolated pairs, most ACs (>90%) are formed by groups of structural analogs with varying potency, creating coordinated clusters that reveal more SAR information than isolated pairs [67]. These can be effectively represented and analyzed as AC networks.
Why Activity Cliffs Challenge Machine Learning Models
The fundamental challenge posed by activity cliffs stems from their violation of the smoothness assumption that underlies many machine learning algorithms. Specifically:
Latent Space Organization: GNNs and other structure-based models naturally embed structurally similar molecules close together in latent space. When these structurally similar molecules have divergent activities, it creates conflicting signals during model training [66].
Data Scarcity Amplification: The problem is particularly acute in low-data regimes common in drug discovery, where models have insufficient examples to learn the complex, non-linear relationships that give rise to ACs [2].
Performance Discrepancies: Studies consistently show that QSAR models experience significant performance drops when predicting "cliffy" compounds involved in ACs compared to non-cliffy compounds [69]. This performance gap persists across model architectures, including highly nonlinear deep learning models.
Table 1: Experimental Evidence of Activity Cliff Impact on Model Performance
| Study | Finding | Implication |
|---|---|---|
| van Tilborg et al. [69] | Descriptor-based QSAR methods outperform complex deep learning models on "cliffy" compounds | Challenges assumption that deep learning automatically solves AC problems |
| ACANet Analysis [66] | Standard GNNs show 31.4% lower label coherence in latent space on BRAF dataset | Demonstrates fundamental architectural limitations |
| Sheridan et al. [69] | AC density strongly predictive of dataset modelability | ACs as key determinant of prediction feasibility |
Modern Approaches for Activity Cliff Mitigation
AC-Informed Contrastive Learning (ACANet)
The AC-awareness (ACA) framework introduces a novel inductive bias designed specifically to enhance molecular representation learning for activity modeling [66]. This approach jointly optimizes metric learning in the latent space and task performance in the target space, making models more sensitive to ACs.
The core innovation is the ACA loss function, which combines a standard regression loss (MAE or MSE) with a Triplet Soft Margin (TSM) loss:
The TSM loss operates on High-Value Activity Cliff Triplets (HV-ACTs), which consist of an anchor compound (A), a positive compound (P) with similar activity, and a negative compound (N) with dissimilar activity. During training, conditional ACTs are mined using two cliff cut-off parameters—cliff lower (cl) and cliff upper (cu)—applied to the activity labels, and only HV-ACTs (those with triplet loss values greater than zero) are used for TSM loss calculation in each batch [66].
Diagram 1: ACANet Architecture - Integrating activity cliff awareness through combined regression and triplet loss
Experimental results across 39 benchmark datasets demonstrate that AC-informed representations consistently outperform standard models in bioactivity prediction for both regression and classification tasks [66]. On low-sample size, narrow scaffold datasets representative of early-stage drug discovery, models with AC-awareness improved label coherence in latent space by 31.4% and showed average performance improvements of 7.54% and 21.6% over baseline models using MAE and MSE regression losses, respectively [66].
Reinforcement Learning-Guided Contrastive Learning (RL-GCL)
RL-GCL represents another advanced approach that addresses the limitations of standard contrastive learning methods, which often treat molecules as generic graphs and ignore molecular significance during augmentation [70]. This method leverages reinforcement learning to generate "hard" molecular augmentations that enrich contrastive learning.
The key innovation is a specialized reward function that considers both molecular similarity and label dissimilarity for positive sample pairs:
This ensures that generated molecular graph augmentations remain consistent with anchor labels while differing in molecular similarity, creating challenging positive samples that enhance model robustness [70]. The approach incorporates label information into the contrastive loss function, enabling more accurate discrimination between positive and negative samples while mitigating bias from false negatives.
Table 2: Comparison of Activity Cliff Mitigation Approaches
| Method | Core Mechanism | Key Components | Reported Performance Gains |
|---|---|---|---|
| ACANet [66] | AC-informed contrastive learning | ACA loss, Triplet Soft Margin, HV-ACT mining | 7.16-21.6% improvement across 39 datasets |
| RL-GCL [70] | RL-guided hard augmentation | Multi-objective reward, supervised contrastive loss | Outperforms SOTA baselines on 8 MoleculeNet tasks |
| Image-Based CNN [68] | Compound pair image analysis | Molecular image concatenation, Grad-CAM visualization | High accuracy in AC/Non-AC classification |
| ACS [2] | Multi-task learning optimization | Adaptive checkpointing, task specialization | Effective with as few as 29 labeled samples |
Multi-Task Learning with Adaptive Checkpointing (ACS)
For scenarios with limited labeled data, Adaptive Checkpointing with Specialization (ACS) provides an effective framework for mitigating negative transfer in multi-task learning while leveraging correlations among related molecular properties [2].
The ACS method combines a shared, task-agnostic GNN backbone with task-specific trainable heads, adaptively checkpointing model parameters when negative transfer signals are detected [2]. This design promotes inductive transfer among sufficiently correlated tasks while protecting individual tasks from deleterious parameter updates, making it particularly valuable in ultra-low data regimes.
During training, the validation loss of every task is monitored, and the best backbone-head pair is checkpointed whenever a task's validation loss reaches a new minimum. This ensures each task ultimately obtains a specialized backbone-head pair optimized for its specific characteristics [2].
Experimental Protocols and Implementation Guidelines
Implementing AC-Informed Contrastive Learning
Protocol 1: ACANet Implementation for Molecular Property Prediction
Data Preparation:
- Curate dataset with standardized activity measurements (pKi, IC50, etc.)
- Generate Matched Molecular Pairs (MMPs) using algorithmic fragmentation
- Apply substituent size restrictions (max 13 non-hydrogen atoms) to limit to structural analogs [68]
- Label MMP-cliffs based on potency threshold (typically ΔpKi ≥ 2.0)
Model Architecture:
- Implement GNN backbone (GIN, GCN, or MPNN)
- Add parallel branches for regression output and latent space embedding
- Configure triplet sampling mechanism for HV-ACT mining
Training Procedure:
- Initialize standard regression loss (MAE/MSE)
- Mine conditional ACTs using cliff lower (cl) and cliff upper (cu) parameters
- Compute TSM loss only on HV-ACTs with loss values > 0
- Combine losses: Total Loss = Regression Loss + α * TSM Loss
- Gradually adjust α to balance structural and activity learning
Evaluation:
- Assess standard regression metrics on test set
- Evaluate AC-sensitivity using cliff compound subsets
- Analyze latent space organization using dimensionality reduction
Protocol 2: RL-GCL Implementation for Molecular Representation Learning
Reinforcement Learning Environment Setup:
- Define state space: current molecular graph
- Define action space: valid molecular modifications
- Configure reward function incorporating molecular similarity and label consistency
Augmentation Generation:
- Use RL agent to generate hard positive samples
- Ensure augmentations maintain label invariance while reducing structural similarity
- Apply validity checks using chemical rules
Contrastive Learning:
- Implement supervised contrastive loss incorporating label information
- Employ strategy to mitigate false negative bias
- Pre-train model on unlabeled molecular data
Downstream Fine-tuning:
- Apply linear evaluation protocol for property prediction
- Alternatively, use semi-supervised fine-tuning with limited labels
- Validate on molecular property benchmarks
Diagram 2: Comprehensive Workflow for AC-Robust Model Development - From data preparation to model deployment
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools and Resources for AC Research
| Tool/Resource | Type | Primary Function | Application in AC Research |
|---|---|---|---|
| RDKit [68] | Cheminformatics Library | Molecular representation and manipulation | MMP generation, 2D image creation, fingerprint calculation |
| ChEMBL [69] | Bioactivity Database | Repository of curated compound activities | Source of high-confidence activity data for AC analysis |
| MATCHED MOLECULAR PAIRS Algorithm [68] | Computational Method | Systematic identification of analog pairs | Core technology for MMP and MMP-cliff identification |
| Grad-CAM [68] | Visualization Technique | Convolutional feature mapping | Identification of structural features contributing to AC predictions |
| DeepPurpose [71] | DTI Prediction Toolkit | Drug-target interaction modeling | Binding affinity prediction for generated molecules |
| BindingDB [71] | Protein-Ligand Database | Binding affinity measurements | Training data for targeted molecule generation |
Activity cliffs represent both a challenge and an opportunity in molecular property prediction. While they disrupt conventional QSAR modeling approaches, they also encode rich structure-activity relationship information that can be leveraged to build more robust and predictive models. The mitigation strategies outlined in this whitepaper—AC-informed contrastive learning, reinforcement learning-guided augmentation, and specialized multi-task learning—provide powerful frameworks for enhancing model performance in the presence of SAR discontinuities.
Future research directions should focus on developing unified frameworks that combine the strengths of these approaches, improving interpretability to extract chemical insights from AC-aware models, and expanding applications beyond primary activity prediction to ADMET properties and multi-property optimization. As the field progresses, the systematic addressing of activity cliffs will move from being a specialized consideration to a fundamental component of molecular property prediction workflows, ultimately accelerating the discovery of novel therapeutic compounds.
Ensuring Reliability: Evaluation Metrics and Performance Validation
Choosing the Right Evaluation Metrics for Your Task
Selecting the appropriate evaluation metrics is a foundational step in machine learning (ML) research for molecular property prediction. Metrics are not merely tools for reporting final performance; they guide model development, enable meaningful comparisons between algorithms, and ultimately determine whether a model will be useful in practical drug discovery applications. The choice of metric is deeply intertwined with the type of molecular property being predicted, the characteristics of the available data, and the real-world context in which predictions will be deployed.
In molecular property prediction, tasks are generally categorized as either regression (predicting continuous values) or classification (predicting categorical labels). Within these broad categories, specific considerations for molecular data—such as dataset size, label imbalance, and the presence of activity cliffs—further complicate metric selection. A metric that works well for predicting quantum mechanical properties of small molecules may be entirely unsuitable for predicting bioactivity or toxicity endpoints. This guide provides a comprehensive framework for selecting and implementing evaluation metrics tailored to the unique challenges of molecular property prediction, enabling researchers to make informed decisions that align with their scientific objectives.
Taxonomy of Molecular Properties and Corresponding Metrics
Molecular properties span a wide spectrum, from quantum mechanical calculations to complex physiological outcomes. The table below catalogs common property types, their characteristics, and recommended evaluation metrics.
Table 1: Metric Selection Guide for Different Molecular Property Types
| Property Category | Example Tasks | Data Characteristics | Recommended Metrics | Rationale |
|---|---|---|---|---|
| Quantum Mechanics [72] | QM7, QM8, QM9 datasets (e.g., atomization energies, electronic properties) | Calculated properties, high precision, ~10k-100k+ samples | Mean Absolute Error (MAE) [72] | Preferred for its interpretability (same units as property) and robustness. |
| Physical Chemistry [72] | ESOL (solubility), FreeSolv (solvation energy), Lipophilicity | Experimental measurements, smaller datasets (~hundreds to thousands of samples) | Root Mean Squared Error (RMSE) [72] | Penalizes larger errors more heavily, important for practical applications. |
| Biophysics & Physiology [72] | Protein-ligand binding, Blood-Brain Barrier (BBB) penetration | Bioactivity measurements, often binary labels (active/inactive) | Area Under the ROC Curve (AUROC) [72] [73] | Evaluates ranking performance across all classification thresholds. |
| Toxicology & Side Effects [72] [2] | Tox21 (toxicity), SIDER (side effects), ClinTox | Highly imbalanced datasets, where inactive/negative class dominates | Area Under the Precision-Recall Curve (PRC-AUC) [73] | More informative than AUROC for imbalanced data; focuses on the positive class. |
| ADMET [74] | Absorption, Distribution, Metabolism, Excretion, Toxicity | Critical for drug safety, often highly imbalanced | AUROC, PRC-AUC, F1-Score [74] | A combination is often needed to assess different aspects of model utility. |
Special Considerations for Data Characteristics
The nature of your dataset significantly impacts metric reliability:
- Data Scarcity: In ultra-low data regimes (e.g., <100 samples), standard metrics can become unstable and exhibit high variance [2]. Reporting confidence intervals via bootstrapping is essential.
- Activity Cliffs: When small structural changes cause large property changes, models can appear to perform well on standard metrics while failing dramatically on these critical cases [73]. Analyzing metric performance specifically on these subsets is crucial.
- Label Imbalance: For classification, a metric like accuracy is misleading with imbalanced data. A model that always predicts "inactive" can achieve high accuracy but is useless. PRC-AUC is strongly recommended in these scenarios [73].
Detailed Methodologies for Metric Implementation and Evaluation
A robust evaluation protocol requires more than calculating a single metric. This section details the experimental workflows and statistical rigor needed for reliable assessment.
Dataset Splitting Strategies
The method used to split data into training, validation, and test sets profoundly affects the realism of your evaluation. The diagram below illustrates the workflow for selecting and implementing a splitting strategy.
Diagram 1: Workflow for selecting a dataset splitting strategy.
- Random Splitting: Molecules are assigned randomly to training, validation, and test sets. This is the least realistic but useful for initial model validation and ablation studies [72].
- Scaffold Splitting: Molecules are split based on their Bemis-Murcko scaffolds, ensuring that the test set contains structurally novel compounds not seen during training. This is the gold standard for estimating a model's ability to generalize to new chemical series and is highly recommended for most applications [73] [74].
- Temporal Splitting: Data is split based on the date it was collected, simulating a real-world scenario where the model predicts properties for molecules synthesized in the future. This helps avoid inflated performance from evaluating on older, structurally similar molecules [2].
- Stratified Splitting: Ensures that the distribution of the target property (e.g., the ratio of active to inactive compounds) is consistent across all splits. This is particularly important for small or highly imbalanced datasets [72].
Statistical Validation Protocols
To ensure reported metrics are reliable and not the result of statistical noise, follow this rigorous protocol:
- Multiple Runs with Different Seeds: For a given data split, run your model training multiple times (e.g., 10) with different random seeds to account for variability in weight initialization and stochastic optimization. Report the mean and standard deviation of your key metrics [73].
- Multiple Splits: Perform step 1 across multiple different splits of the data (e.g., 3-5 different scaffold splits). This accounts for variability stemming from how the data was partitioned.
- Statistical Significance Testing: When comparing a new model (Model A) against a baseline (Model B), use a paired statistical test. A recommended method is the paired t-test:
- For each of the N data splits, calculate the performance difference for a given metric (e.g.,
AUROC_A - AUROC_B). - Perform a one-sided t-test on these N differences to determine if the improvement of Model A over Model B is statistically significant (typically with a p-value < 0.05).
- For each of the N data splits, calculate the performance difference for a given metric (e.g.,
Advanced Multi-Task Evaluation
When predicting multiple properties simultaneously (Multi-Task Learning or MTL), evaluation becomes more complex. The ACS (Adaptive Checkpointing with Specialization) training scheme provides a robust framework [2].
- Architecture: Use a shared graph neural network (GNN) backbone with task-specific multi-layer perceptron (MLP) heads.
- Checkpointing: During training, monitor the validation loss for each task independently. Checkpoint the best backbone-head pair for a task whenever its validation loss reaches a new minimum.
- Final Evaluation: For each task, use its specialized checkpoint to make predictions on the test set and calculate the relevant metric (e.g., AUROC for classification, RMSE for regression). This approach mitigates "negative transfer," where updates for one task degrade performance on another [2].
The Scientist's Toolkit: Essential Research Reagents
This table details key computational tools and resources required for rigorous evaluation in molecular property prediction.
Table 2: Essential Tools and Resources for Model Evaluation
| Tool / Resource | Type | Primary Function in Evaluation | Key Features |
|---|---|---|---|
| DeepChem [72] | Software Library | Provides standardized implementations of datasets, featurizations, models, and metrics. | Integrated with MoleculeNet; offers data splitting methods and metric calculators. |
| MoleculeNet [72] [75] | Benchmark Suite | Serves as a curated collection of public datasets for standardized benchmarking. | Categorizes datasets (QM, PhysChem, etc.) and recommends metrics and splits. |
| RDKit [73] | Cheminformatics Toolkit | Handles molecular standardization, descriptor calculation, and scaffold-based splitting. | Critical for generating Bemis-Murcko scaffolds for a rigorous train/test split. |
| TDC (Therapeutics Data Commons) [74] | Benchmark Suite | Offers additional datasets, particularly in the ADMET domain, for model validation. | Complements MoleculeNet with a focus on pharmacology-relevant tasks. |
| FGBench [19] | Specialized Dataset | Enables evaluation of models (especially LLMs) on functional group-level reasoning. | Provides fine-grained annotations to assess understanding of structure-property relationships. |
Limitations, Pitfalls, and Future Directions
Despite the framework provided above, researchers must be aware of significant limitations in current benchmarking practices.
- Over-reliance on Problematic Benchmarks: Widely used benchmarks like MoleculeNet contain issues that can invalidate comparisons, including invalid chemical structures, inconsistent stereochemistry, undefined training/test splits, and aggregation of data from inconsistent experimental sources [76]. For example, the BBB dataset in MoleculeNet contains duplicate structures with conflicting labels [76].
- Metric Misalignment with Application: A model might achieve a high AUROC on a toxicity benchmark but still fail in practice. In virtual screening, the true positive rate at a low false positive rate is often more relevant than the overall AUROC, as only a small fraction of a compound library will be tested experimentally [73].
- The Low-Data Regime Challenge: Learnable representations like Graph Neural Networks (GNNs) often struggle with complex tasks under data scarcity [72] [2]. In these cases, simpler models using physics-aware featurizations or molecular fingerprints can be more effective and reliable [72] [73].
Future directions involve developing more rigorous and application-relevant benchmarks. This includes creating datasets with carefully curated and standardized experimental data, defining more clinically relevant evaluation metrics, and placing greater emphasis on a model's performance in low-data regimes and its ability to generalize to truly novel scaffolds.
For researchers and scientists embarking on machine learning (ML) for molecular property prediction, the selection of a dataset splitting strategy is a foundational decision that profoundly impacts the real-world applicability of developed models. This choice determines how well a model's reported performance will translate from benchmark datasets to the true challenge of drug discovery: predicting properties for novel, previously unseen chemical structures. Within this context, the debate between random splitting and scaffold-based splitting represents a critical junction, separating convenient benchmarking from chemically meaningful evaluation. Random splitting, while simple and computationally efficient, often leads to overly optimistic performance estimates by allowing structurally similar molecules to appear in both training and test sets. In contrast, scaffold-based splitting provides a more challenging and realistic assessment by forcing models to predict properties for molecules with entirely different core structures, a scenario frequently encountered in prospective drug discovery campaigns. This technical guide provides an in-depth examination of these strategies, equipping researchers with the knowledge to implement rigorous evaluation protocols that accurately gauge model generalizability within chemical space.
Molecular Representations and Property Prediction
Molecular property prediction seeks to learn a function that maps a chemical structure to a specific property value, a core task in AI-driven drug discovery [77]. The choice of how to represent a molecule computationally is the first critical step in this pipeline, directly influencing model performance and generalization.
- Fixed Representations: These include predefined molecular descriptors and fingerprints. Common examples are 1D/2D descriptors (e.g., molecular weight, polar surface area) and 2D fingerprints like Extended-Connectivity Fingerprints (ECFP), which encode the presence of specific substructural patterns as bit vectors [77].
- SMILES Strings: Simplified Molecular-Input Line-Entry System (SMILES) represents the molecular graph as a linear string of symbols. While computationally efficient, a single molecule can have multiple valid SMILES strings, and this representation inherently ignores structural information [77].
- Molecular Graphs: This representation intuitively depicts atoms as nodes and bonds as edges in a graph. Graph Neural Networks (GNNs) can then operate directly on this structure to learn task-specific representations, capturing rich structural information that is lost in SMILES strings [77] [78].
Despite the advent of sophisticated representation learning models, their performance is often bottlenecked by the quality and size of the datasets used for training and evaluation [77]. A model's ability to generalize depends not just on its architecture but critically on how the data is split to simulate real-world application scenarios.
Dataset Splitting Strategies
The method used to partition a dataset into training, validation, and test sets is paramount for a realistic evaluation of a model's generalizability. The core challenge is to avoid information leakage, where a model performs well on a test set because it has been trained on molecules that are overly similar, thus failing to learn generalizable structure-property relationships [79].
Random Split
The random split is the most straightforward approach, where molecules are assigned randomly to training, validation, and test sets, typically in proportions like 70-80% for training and the remainder for testing and validation [80] [81].
- Mechanism: A random number generator is used to shuffle the dataset and assign molecules to different subsets. This process can be controlled by a random seed to ensure reproducibility [81].
- Advantages: Its primary advantage is simplicity and the conservation of overall label distribution across splits, which is useful for initial model development and debugging.
- Disadvantages: The major drawback is its tendency to create an overly optimistic evaluation. With random splitting, it is common for the test set to contain molecules that are structurally very similar to those in the training set. The model can then achieve high performance by leveraging these similarities rather than by learning robust, generalizable patterns [80] [78]. This provides a poor approximation of the real-world scenario where models are applied to genuinely novel chemical scaffolds [78].
Scaffold Split
The scaffold split, also known as the Bemis-Murcko scaffold split, addresses the limitations of random splitting by explicitly partitioning data based on the core molecular structure.
- Mechanism: The Bemis-Murcko algorithm is applied to each molecule to iteratively remove monovalent atoms (typically side chains and functional groups), leaving behind the core scaffold [80]. Molecules are then grouped by these scaffolds, and the splitting ensures that all molecules sharing an identical scaffold are assigned to the same subset (training, validation, or test) [80] [81]. This guarantees that the model is tested on scaffolds it has never encountered during training.
- Advantages: This method provides a more challenging and realistic benchmark of a model's ability to generalize across distinct chemical classes, which is crucial for hit identification in novel chemical series [82] [78].
- Disadvantages and Nuances: A key challenge arises when two highly similar molecules, differing by only a single atom, are assigned different scaffolds and thus end up in different splits [80]. While this tests true generalization, it can sometimes make the test set artificially difficult. Furthermore, the distribution of scaffold sizes can lead to imbalanced training and test set sizes [80].
Table 1: Comparison of Core Dataset Splitting Strategies
| Strategy | Partitioning Principle | Key Advantages | Key Limitations |
|---|---|---|---|
| Random Split | Random assignment of molecules | Simple to implement; preserves label distribution | Overly optimistic performance; poor simulation of real-world generalization |
| Scaffold Split | Grouping by Bemis-Murcko core structure | Tests generalization to novel scaffolds; more realistic benchmark | Can be overly challenging; test/train set sizes may vary |
| Butina Split | Clustering by molecular fingerprint similarity | Groups by overall structural similarity, not just core | Clustering quality depends on parameters; similar scaffolds can be in different clusters |
| UMAP Split | Clustering in a low-dimensional, non-linear projection | Can achieve high cluster separation and molecular dissimilarity | Complex workflow; requires tuning (e.g., number of clusters) [80] [82] |
Advanced and Alternative Splitting Strategies
Beyond random and scaffold splits, several other methods have been developed to introduce varying levels of rigor and realism into model evaluation.
- Butina Split: This method uses the Butina clustering algorithm on molecular fingerprints (like Morgan fingerprints) to group molecules by overall structural similarity. Molecules within the same cluster are kept together in the same data split [80] [82]. It aims to create more chemically distinct splits than random splitting.
- UMAP Split: This is a more recent and rigorous approach. It involves projecting molecular fingerprints into a low-dimensional space using the UMAP algorithm and then clustering the projected coordinates. This method has been shown to create splits with high inter-cluster molecular dissimilarity, providing a more challenging and realistic benchmark than scaffold or Butina splits [82]. The number of clusters is a key parameter, with higher numbers (e.g., >35) leading to more stable test set sizes [80].
- Cold-Split & Time Split: For tasks involving multiple entities (e.g., drug-target interaction), a cold split ensures that specific entities (like a particular drug) are entirely absent from the training set [81]. A time-based split, which is considered ideal for mimicking real-world deployment, involves training on older data and testing on newer data, but it requires timestamped data that is often unavailable [80].
Experimental Protocols and Implementation
Implementing rigorous dataset splits requires careful methodology. Below is a detailed protocol for a scaffold split, one of the most common non-trivial strategies.
Detailed Protocol: Implementing a Scaffold Split
Objective: To partition a molecular dataset into training and test sets such that no molecular scaffold is shared between the two sets, thereby rigorously assessing model generalizability.
Materials & Reagents:
- A dataset containing molecular structures (e.g., as SMILES strings) and associated property labels.
- A computing environment with RDKit installed (required for scaffold generation and fingerprint calculation) [80] [81].
- TDC (Therapeutics Data Commons) library or the
splitoPython package, which provide built-in functions for various splitting strategies [81] [83].
Procedure:
- Data Preprocessing: Standardize the molecular structures from the dataset (e.g., neutralize charges, remove solvents) using a toolkit like RDKit to ensure consistency.
- Scaffold Generation: For each molecule in the dataset, generate its Bemis-Murcko scaffold using the RDKit function
GetScaffoldForMol. This process removes side chains and returns the core ring system with linker atoms. - Group Assignment: Assign every molecule to a group based on its canonical scaffold representation. Each unique scaffold defines a distinct group.
- Stratified Splitting: Use a splitting function that respects these group assignments. The
GroupKFoldorGroupKFoldShufflemethod from libraries like scikit-learn or TDC is ideal for this [80]. This function will ensure that all molecules belonging to the same scaffold group are allocated to the same data fold. - Train-Test Partition: Execute the split, typically using an 80/20 ratio for train and test sets. The indices for the training and test sets are returned.
- Validation Set: Further split the training set (e.g., using an 80/10/10 ratio for train/validation/test) using a nested group split or a random split, depending on the need for rigor during hyperparameter tuning.
Validation of the Split:
- It is crucial to verify the chemical dissimilarity between the training and test sets. A qualitative method is to visualize the chemical space using a projection like t-SNE or PCA, coloring points by their dataset split. A successful scaffold split will show intermingled red and blue points, indicating shared chemical space but distinct scaffolds [80].
- A more quantitative method, inspired by Sheridan's work, is to calculate the maximum Tanimoto similarity between each test set molecule and its nearest neighbor in the training set. A lower average maximum similarity indicates a more successful and challenging split [80].
The following workflow diagram illustrates the key steps in this protocol.
Impact on Model Performance and Evaluation
The choice of splitting strategy has a dramatic and quantifiable impact on the perceived performance of ML models, a fact that is sometimes overlooked in the literature.
Quantitative Performance Gaps
Extensive benchmarking studies reveal a consistent pattern: model performance metrics are significantly higher when evaluated with random splits compared to more rigorous scaffold or cluster-based splits. A large-scale study training over 62,000 models found that representation learning models exhibit limited performance in most realistic evaluation settings, underscoring the importance of dataset size and split rigor [77].
Another study involving 8,400 models across 60 cancer cell line datasets showed a clear hierarchy of difficulty based on the splitting method. The reported performance, especially for complex deep learning models, was highest for random splits, followed by scaffold splits, then Butina splits, with UMAP-based clustering splits yielding the most challenging and realistic evaluation [82]. This demonstrates that scaffold splits are not the ultimate challenge; more advanced splitting methods can reveal further performance drops.
Implications for Virtual Screening (VS)
The impact of data splitting is particularly critical in the context of virtual screening. The most commonly used metric in benchmarks, the ROC AUC, has been criticized as a suboptimal metric for VS. This is because ROC AUC considers the entire ranking of molecules, whereas in VS, the primary interest is in the early recognition of a very small number of top-ranked hits [82]. Therefore, a rigorous data split combined with a VS-relevant metric like hit rate at a very early cutoff (e.g., the top 100 molecules) is essential for a realistic assessment of a model's utility in a drug discovery pipeline [82].
Table 2: Impact of Splitting Strategy on Model Evaluation and Real-World Utility
| Evaluation Aspect | Random Split | Scaffold Split | Advanced Splits (e.g., UMAP) |
|---|---|---|---|
| Reported Performance | Inflated, overly optimistic | More conservative, challenging | Most conservative, highly challenging |
| Generalization Test | Poor; tests on similar molecules | Good; tests on novel scaffolds | Excellent; tests on highly dissimilar chemotypes |
| Relevance to VS | Low; does not reflect real-world library diversity | Moderate to High | High; better mimics diversity of screening libraries |
| Recommended Metric | ROC AUC can be misleading | Early recognition metrics (e.g., Hit Rate @ k) are more informative [82] | Early recognition metrics (e.g., Hit Rate @ k) are critical [82] |
To implement the strategies discussed in this guide, researchers can leverage a suite of powerful software tools and libraries.
Table 3: Essential Tools and Resources for Rigorous Data Splitting
| Tool / Resource | Type | Key Functionality | Application Note |
|---|---|---|---|
| RDKit | Cheminformatics Library | Bemis-Murcko scaffold generation, molecular fingerprint calculation, descriptor computation. | The foundational tool for most custom splitting implementations and chemical informatics work [80]. |
| Therapeutics Data Commons (TDC) | Python Library/Platform | Provides standardized data loaders and built-in split functions (random, scaffold, cold) for benchmark datasets. | Excellent for getting started quickly with standardized benchmarks and ensuring comparability with published results [81]. |
| scikit-learn | Machine Learning Library | Provides GroupKFold and related functions for implementing group-based splits like scaffold split. |
Essential for creating custom data splitting pipelines integrated into an ML workflow [80]. |
| DataSAIL | Python Package | Specialized tool for creating splits that minimize information leakage for biomedical data (1D and 2D). | Useful for complex scenarios, such as splitting drug-target interaction data to ensure no protein or drug leaks between sets [79]. |
| splito | Python Library | A dedicated library for implementing various chemical data splitting strategies. | Simplifies the process of applying multiple splitting methods with a consistent API [83]. |
For researchers and professionals in drug development, the message is clear: the default use of random data splitting is insufficient for developing ML models that are truly robust and generalizable. While scaffold splitting represents a significant step forward by enforcing a separation of core chemical structures between training and test data, it is not the final word in rigorous evaluation. Emerging methods like UMAP-based clustering and tools like DataSAIL offer even more robust ways to simulate the profound chemical diversity encountered in real-world virtual screening libraries. By adopting these more challenging splitting strategies and aligning evaluation metrics with practical goals like early hit identification, the field can bridge the gap between impressive benchmark performance and successful prospective drug discovery. The path to reliable molecular property prediction begins with a rigorous and realistic approach to evaluating our models.
Benchmarking Model Performance Across Representations
Molecular property prediction stands as a fundamental task in computer-aided drug discovery, where the accurate prediction of molecular properties from chemical structure can significantly accelerate early-stage research and development. The selection of an appropriate molecular representation—the method of encoding chemical structures into a computational model—profoundly impacts the accuracy, generalizability, and ultimate utility of machine learning (ML) models [84] [73]. Despite the proliferation of novel representation learning techniques, systematic benchmarking reveals that no single representation consistently outperforms others across diverse tasks and datasets [73] [85]. This technical guide provides a structured framework for benchmarking model performance across molecular representations, enabling researchers to make informed decisions tailored to their specific property prediction objectives, data constraints, and computational resources.
The performance of molecular machine learning models is intrinsically linked to the choice of representation. Different representations capture varying aspects of molecular structure and chemistry, leading to significant differences in model generalization, especially on out-of-distribution (OOD) data [3] [85]. Recent large-scale benchmarking efforts demonstrate that even state-of-the-art deep learning models exhibit OOD errors approximately three times larger than their in-distribution performance [3]. This performance variability underscores the critical need for rigorous, standardized benchmarking protocols to guide representation selection and model development in scientific and industrial applications.
Molecular Representation Paradigms
Molecular representations can be broadly categorized into traditional fixed representations and learned representations. Each paradigm offers distinct advantages and limitations, which must be evaluated within the context of specific prediction tasks.
Traditional Fixed Representations
Traditional representations rely on predefined, rule-based feature extraction methods developed through decades of cheminformatics research [84].
- Molecular Descriptors: These are numerical values quantifying physical or chemical properties (e.g., molecular weight, hydrophobicity, topological indices) [73]. RDKit2D descriptors provide approximately 200 predefined molecular features, offering a comprehensive baseline for model performance [73].
- Molecular Fingerprints: These encode substructural information as binary strings or numerical vectors [84]. Extended-Connectivity Fingerprints (ECFP), based on the Morgan algorithm, are the de facto standard circular fingerprint, capturing molecular features through iterative atom environment updates [73]. Other fingerprint types include Molecular ACCess System (MACCS) keys and atom pair fingerprints [73].
Learned Representations
Modern AI-driven approaches employ deep learning to automatically learn continuous, high-dimensional feature embeddings directly from data [84].
- Language Model-Based Representations: Inspired by natural language processing, these methods treat molecular sequences (e.g., SMILES, SELFIES) as chemical language [84]. Models such as Transformers and BERT tokenize these strings and process them to learn representations that capture sequential patterns [84].
- Graph-Based Representations: These intuitively represent molecules as graphs with atoms as nodes and bonds as edges [73]. Graph Neural Networks (GNNs), including Graph Convolutional Networks (GCNs), learn representations by aggregating information from neighboring atoms, directly capturing structural connectivity [73] [86].
- 3D Structure-Based Representations: These incorporate spatial atomic coordinates, either as 3D molecular graphs or 3D molecular grids, providing critical information about molecular conformation and shape [86].
Table 1: Comparison of Major Molecular Representation Types
| Representation Type | Key Examples | Advantages | Limitations |
|---|---|---|---|
| Molecular Descriptors | RDKit2D, PhysChem descriptors [73] | Computationally efficient, interpretable, strong baseline performance | Limited to predefined features, may miss complex structural patterns |
| Molecular Fingerprints | ECFP, MACCS keys [73] | Effective for similarity search, works well with traditional ML | Hand-crafted nature, limited extrapolation to novel chemistries |
| Language Model-Based | SMILES-BERT, Transformer-based models [84] | No need for expert-designed features, can learn from large unlabeled corpora | SMILES syntax limitations, may generate invalid structures |
| Graph-Based | GCN, GNN, Message Passing Networks [73] [86] | Directly captures molecular topology, state-of-the-art on many tasks | Computationally intensive, requires careful hyperparameter tuning |
| 3D Structure-Based | 3D-CNN, Geometric GNNs [86] | Captures spatial and conformational information, crucial for quantum properties | Requires 3D conformation data, which may be limited or computationally expensive to generate |
Benchmarking Methodology and Experimental Design
Robust benchmarking requires standardized datasets, appropriate data splitting strategies, meaningful evaluation metrics, and controlled experimental protocols.
Standardized Datasets and Data Splitting
The MoleculeNet benchmark provides a curated collection of datasets spanning quantum mechanics, physical chemistry, biophysics, and physiology, enabling direct comparison across studies [72]. Key datasets include:
- Quantum Mechanics: QM7, QM8, QM9 for predicting quantum mechanical properties [72].
- Physical Chemistry: ESOL (solubility), FreeSolv (hydration free energy), Lipophilicity [72].
- Physiology: Tox21 (toxicity), MUV (virtual screening), HIV [72].
Critical to meaningful benchmarking is the data splitting method. Random splitting, common in ML, is often inappropriate for chemical data due to the risk of data leakage between training and test sets [72]. Scaffold splitting, which separates compounds based on their core molecular frameworks, provides a more challenging and realistic assessment of a model's ability to generalize to novel chemotypes [73] [72]. Time-based splits may be used for datasets collected over time to simulate real-world deployment conditions [72].
Evaluation Metrics and Statistical Rigor
Evaluation metrics must align with the task type and practical objectives:
- Regression Tasks: Mean Absolute Error (MAE), Root Mean Squared Error (RMSE) [72].
- Classification Tasks: Area Under the Receiver Operating Characteristic Curve (AUROC), Area Under the Precision-Recall Curve (AUPRC) [73].
Statistical rigor is paramount. Single runs with arbitrary data splits can produce misleading results due to inherent variability [73]. Benchmarking should incorporate multiple random seeds (minimum 3-10) with different dataset initializations, and results should be reported as mean ± standard deviation to account for this variability [73].
Diagram 1: Benchmarking Workflow
Quantitative Benchmarking Results and Performance Analysis
Large-scale systematic studies provide critical insights into the relative performance of different representations across diverse chemical tasks.
Performance Across Representation Types
A comprehensive study training over 62,000 models revealed that representation learning models (e.g., GNNs, transformers) often show limited performance advantages over traditional fixed representations on many benchmark tasks [73]. The performance hierarchy is highly task-dependent, with no single representation achieving dominance across all chemical domains [73] [85].
Table 2: Performance Comparison Across Representations on Select MoleculeNet Datasets (MAE/RMSE)
| Dataset (Task Type) | ECFP + RF | RDKit2D + MLP | GCN | Transformer | Top Performer |
|---|---|---|---|---|---|
| ESOL (Solubility Regression) | 0.88 [73] | 0.90 [73] | 0.86 [73] | 1.03 [73] | GCN |
| FreeSolv (Hydration Free Energy) | 1.82 [73] | 1.90 [73] | 1.75 [73] | 2.15 [73] | GCN |
| Lipophilicity (Octanol/Water Distribution) | 0.65 [73] | 0.69 [73] | 0.63 [73] | 0.75 [73] | GCN |
| Tox21 (Toxicity Classification) | 0.811 (AUROC) [73] | 0.805 (AUROC) [73] | 0.821 (AUROC) [73] | 0.798 (AUROC) [73] | GCN |
Impact of Dataset Size and Activity Cliffs
Dataset size significantly influences the relative performance of representation types. Traditional fixed representations (e.g., ECFP) frequently outperform learned representations in low-data regimes (typically < 10,000 samples) due to their lower model complexity and reduced risk of overfitting [73]. As dataset size increases (> 50,000 samples), learned representations such as GNNs begin to demonstrate stronger performance by leveraging their capacity to identify complex, non-linear structure-property relationships [73].
The presence of activity cliffs—pairs of structurally similar molecules with large differences in property values—significantly degrades model prediction accuracy for all representation types [73] [85]. Quantifying dataset difficulty using indices like the Roughness Index (ROGI) or the Structure-Activity Landscape Index (SALI) provides a priori insight into expected model performance, with higher roughness values correlating with increased prediction error [85].
Advanced Considerations: Out-of-Distribution Generalization and Representation Topology
The OOD Generalization Challenge
A critical frontier in molecular ML is improving model performance on out-of-distribution data, essential for discovering novel molecules that extend beyond known chemical space [3]. The BOOM (Benchmarking Out-Of-distribution Molecular property predictions) benchmark demonstrates that current models struggle with OOD generalization, with even top-performing models exhibiting average OOD errors three times larger than in-distribution errors [3]. Key findings include:
- Deep learning models with high inductive bias can perform well on OOD tasks with simple, specific properties [3].
- Current chemical foundation models, despite promises of transfer and in-context learning, do not yet show strong OOD extrapolation capabilities [3].
- Factors such as data generation, pre-training strategies, and hyperparameter optimization significantly impact OOD performance [3].
Topological Analysis of Representations
Emerging research explores the connection between the topological characteristics of a representation's feature space and its resulting ML performance [85]. The TopoLearn model demonstrates that topological descriptors derived from persistent homology can predict the effectiveness of representations on specific datasets [85]. This approach offers a novel, more principled method for representation selection, potentially reducing reliance on exhaustive empirical testing.
Diagram 2: Representation Topology & Performance
Practical Implementation and Research Reagents
Successful implementation requires a toolkit of software libraries, datasets, and computational resources.
Table 3: Essential Research Reagents for Molecular Property Prediction Benchmarking
| Tool Category | Specific Tool / Resource | Function and Purpose |
|---|---|---|
| Cheminformatics Libraries | RDKit [73], Open Babel [86] | Generate traditional representations (descriptors, fingerprints), handle molecular I/O, and perform basic cheminformatics operations. |
| Deep Learning Frameworks | DeepChem [72], PyTorch Geometric, DGL-LifeSci [73] | Provide implementations of GNNs and other deep learning models specifically designed for molecular data. |
| Benchmark Datasets | MoleculeNet [72], ChEMBL [73] | Curated, standardized datasets for training and evaluating models across diverse molecular tasks. |
| Hyperparameter Optimization | Optuna, Scikit-Optimize | Automate the search for optimal model parameters, which is critical for fair comparison between representations. |
| Topological Analysis | TopoLearn [85], Giotto-tda | Analyze the topological features of representation spaces to predict model performance and understand representation quality. |
Recommended Experimental Protocol
- Problem Formulation: Clearly define the property prediction task (regression/classification) and identify relevant chemical domains.
- Dataset Curation: Select appropriate benchmark datasets (e.g., from MoleculeNet) or curate proprietary data. Perform rigorous cleaning and standardization.
- Representation Generation: Generate a diverse set of representations spanning traditional (ECFP, descriptors) and learned (graph, sequence) types.
- Model Training with Cross-Validation: Implement multiple models aligned with each representation type. Use a consistent hyperparameter optimization protocol across all models. Employ scaffold splitting for a realistic assessment of generalization.
- Evaluation and Analysis: Evaluate models on held-out test sets using multiple relevant metrics. Perform statistical significance testing on results. Analyze failures and correlate performance with dataset characteristics (e.g., ROGI, SARI).
Benchmarking model performance across molecular representations is not a one-time activity but an ongoing process essential for advancing molecular property prediction. The field has matured beyond simply claiming superiority of a new model on a few selected datasets. Robust benchmarking requires standardized protocols, rigorous statistical analysis, and honest acknowledgment of limitations, particularly concerning out-of-distribution generalization [3] [73]. By adopting the systematic framework outlined in this guide—encompassing diverse representation paradigms, controlled experimental methodologies, and advanced topological analysis—researchers can build more effective, reliable, and generalizable molecular machine learning systems, ultimately accelerating scientific discovery and drug development.
Statistical Rigor in Model Evaluation and Reporting
In machine learning-based molecular property prediction, statistical rigor separates scientifically valid findings from spurious results. The high-stakes nature of drug discovery—where models influence decisions impacting years of research and billions of dollars—demands rigorous evaluation practices [87]. Despite technical advancements, evaluation methodologies often suffer from statistical shortcomings, including inappropriate metric selection, inadequate dataset splitting, and insufficient statistical testing [73] [88]. This guide provides researchers with a comprehensive framework for statistically rigorous model evaluation and reporting, specifically contextualized for molecular property prediction. By adopting these practices, researchers can enhance the reliability, reproducibility, and practical utility of their predictive models, ultimately accelerating robust AI-driven molecular discovery.
Foundational Evaluation Metrics for Molecular Property Prediction
Selecting appropriate evaluation metrics is paramount for accurate model assessment. Metrics must align with both the machine learning task type (classification, regression) and the specific requirements of molecular property prediction contexts (e.g., handling class imbalance, confidence calibration).
Classification Metrics
Molecular property prediction frequently involves classification tasks, such as predicting toxicity, activity, or metabolic stability. The following metrics are essential for comprehensive assessment [89] [90] [88].
Table 1: Key Classification Metrics for Molecular Property Prediction
| Metric | Formula | Interpretation | Molecular Application Context |
|---|---|---|---|
| Accuracy | (TP+TN)/(TP+TN+FP+FN) | Overall correctness | Can be misleading for imbalanced datasets (e.g., few active compounds) [89] |
| Precision | TP/(TP+FP) | Proportion of correct positive predictions | Critical when false positives are costly (e.g., virtual screening prioritization) [89] |
| Recall (Sensitivity) | TP/(TP+FN) | Proportion of actual positives correctly identified | Essential when missing positive cases is undesirable (e.g., toxicity prediction) [89] [88] |
| Specificity | TN/(TN+FP) | Proportion of actual negatives correctly identified | Important for specificity-driven tasks (e.g., minimizing false alarms in high-throughput screening) [88] |
| F1-Score | 2×(Precision×Recall)/(Precision+Recall) | Harmonic mean of precision and recall | Balanced measure when seeking equilibrium between precision and recall [89] [90] |
| AUC-ROC | Area under ROC curve | Model's ability to distinguish between classes | Threshold-independent measure; useful for overall performance assessment [89] [88] |
| Matthews Correlation Coefficient (MCC) | (TP×TN-FP×FN)/√((TP+FP)(TP+FN)(TN+FP)(TN+FN)) | Correlation between observed and predicted | More reliable for imbalanced datasets than accuracy [88] |
| Log Loss | -1/N ∑[yᵢlog(pᵢ)+(1-yᵢ)log(1-pᵢ)] | Uncertainty of predictions based on probability | Penalizes overconfident incorrect predictions; useful for probabilistic interpretation [89] |
For multi-class molecular classification problems (e.g., predicting multiple toxicity endpoints), macro-averaging computes the metric independently for each class and then takes the average, giving equal weight to all classes, while micro-averaging aggregates contributions of all classes, favoring more frequent classes [88].
Regression Metrics
Predicting continuous molecular properties (e.g., solubility, partition coefficients, binding affinities) requires specialized regression metrics [89] [37].
Table 2: Key Regression Metrics for Molecular Property Prediction
| Metric | Formula | Interpretation | Molecular Application Context |
|---|---|---|---|
| Mean Absolute Error (MAE) | (1/N)∑⎮yᵢ-ŷᵢ⎮ | Average absolute difference | Provides intuitive, linearly interpretable error magnitude [89] |
| Mean Squared Error (MSE) | (1/N)∑(yᵢ-ŷᵢ)² | Average squared difference | Heavily penalizes larger errors; sensitive to outliers [89] |
| Root Mean Squared Error (RMSE) | √[(1/N)∑(yᵢ-ŷᵢ)²] | Root of average squared difference | In same units as target variable; emphasizes larger errors [89] |
| R-squared (R²) | 1 - [∑(yᵢ-ŷᵢ)²/∑(yᵢ-ȳ)²] | Proportion of variance explained | Indicates goodness-of-fit; range (-∞, 1] with 1 being perfect fit [89] |
| Root Mean Squared Logarithmic Error (RMSLE) | √[(1/N)∑(log(yᵢ+1)-log(ŷᵢ+1))²] | Root of average squared logarithmic error | Penalizes underestimates more than overestimates; useful for properties spanning orders of magnitude [89] |
Experimental Design for Statistical Rigor
Dataset Splitting Strategies
Proper dataset splitting is crucial for reliable performance estimation and preventing data leakage [73].
Dataset Splitting Workflow
The most critical consideration in molecular property prediction is temporal validation versus random splitting. Random splits often overestimate performance because structurally similar molecules may appear in both training and test sets. Temporal splits, where models are trained on older data and tested on newer data, better reflect real-world deployment scenarios [2]. For molecular data, scaffold splitting groups molecules by their core Bemis-Murcko scaffolds, ensuring that different structural classes are separated between training and test sets, providing a more challenging and realistic evaluation [2] [73].
Statistical Testing for Model Comparison
When comparing machine learning models, observed performance differences must be statistically validated rather than assumed meaningful based on point estimates alone [88].
Methodology for Statistical Comparison:
Generate Multiple Performance Estimates: Use k-fold cross-validation (typically k=5 or 10) or repeated random splits to obtain multiple performance values for each model, rather than a single train-test split [88].
Select Appropriate Statistical Test:
- Paired t-test: Applicable when comparing two models across multiple datasets or cross-validation folds, assuming performance differences are normally distributed.
- Wilcoxon signed-rank test: Non-parametric alternative to paired t-test; does not assume normality of differences.
- Friedman test with post-hoc analysis: For comparing multiple models across multiple datasets, followed by pairwise comparisons if overall significant differences are detected [88].
Correct for Multiple Testing: When performing multiple pairwise comparisons, apply corrections such as Bonferroni or Holm-Bonferroni to control family-wise error rate.
Report Effect Sizes: Beyond statistical significance, report confidence intervals and effect sizes to indicate practical significance of observed differences.
Statistical Testing Decision Process
Advanced Considerations in Molecular Property Prediction
Addressing Data Scarcity and Imbalance
Molecular property datasets often suffer from severe data scarcity and class imbalance, particularly for rare endpoints or novel chemical series [2] [91].
Advanced Techniques:
Multi-Task Learning (MTL): MTL improves generalization by jointly learning multiple related properties, leveraging correlations between tasks. However, performance can be degraded by negative transfer when tasks are not sufficiently related or training datasets are imbalanced [2].
Adaptive Checkpointing with Specialization (ACS): This advanced MTL approach combines a shared task-agnostic backbone with task-specific heads, adaptively checkpointing model parameters when negative transfer is detected. ACS has demonstrated accurate predictions with as few as 29 labeled samples in sustainable aviation fuel property prediction [2].
Few-Shot Learning Approaches: Meta-learning frameworks enable models to generalize from limited examples by learning across multiple related tasks [91]. Context-informed few-shot learning incorporates both property-shared and property-specific molecular features through heterogeneous meta-learning [91].
Data Consistency Assessment
Data heterogeneity and distributional misalignments pose critical challenges in molecular property prediction, particularly when integrating public datasets [92].
AssayInspector Methodology:
- Statistical Comparison: Applies Kolmogorov-Smirnov tests for regression tasks and Chi-square tests for classification to compare endpoint distributions across data sources [92].
- Chemical Space Analysis: Uses Tanimoto similarity (for fingerprints) or Euclidean distance (for descriptors) with UMAP visualization to detect dataset coverage differences [92].
- Annotation Consistency Check: Identifies conflicting property annotations for shared molecules across different datasets [92].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools for Molecular Property Prediction Research
| Tool/Category | Specific Examples | Function | Application Context |
|---|---|---|---|
| Molecular Representations | ECFP fingerprints, RDKit 2D descriptors, SMILES strings, Molecular graphs | Encode molecular structure in machine-readable format | ECFP6 commonly used for traditional ML; graph representations for GNNs [73] [87] |
| Graph Neural Networks | GIN, EGNN, Graphormer | Learn directly from molecular graph structure | GIN for 2D topology; EGNN for 3D geometric properties; Graphormer for global attention [37] |
| Multi-Task Learning Frameworks | Adaptive Checkpointing with Specialization (ACS) | Mitigate negative transfer in imbalanced multi-task settings | Particularly effective in ultra-low data regimes [2] |
| Data Consistency Assessment | AssayInspector | Identify dataset discrepancies before model training | Critical when integrating heterogeneous data sources [92] |
| Benchmark Datasets | MoleculeNet, TDC, ChEMBL | Standardized benchmarks for model comparison | Ensure fair evaluation; be aware of dataset limitations and relevance to real-world applications [73] [92] |
| Statistical Testing Packages | SciPy, scikit-posthocs | Perform statistical comparisons of model performance | Essential for validating claimed improvements [88] |
Best Practices for Reporting Results
To ensure reproducibility and facilitate proper interpretation, adhere to these reporting standards:
- Complete Metric Reporting: Report multiple metrics to provide a comprehensive view (e.g., both precision and recall, not just accuracy) [73] [88].
- Dataset Characterization: Report dataset size, class distribution, and splitting methodology (including specific scaffolds for scaffold splits) [73].
- Uncertainty Quantification: Report performance as mean ± standard deviation across multiple runs rather than single point estimates [88].
- Statistical Significance: Report p-values and confidence intervals for model comparisons, with appropriate multiple testing corrections [88].
- Code and Data Availability: Share preprocessing code, model configurations, and data splitting details to enable exact reproduction [73].
By implementing these statistically rigorous evaluation and reporting practices, researchers in molecular property prediction can produce more reliable, reproducible, and practically useful models that genuinely advance drug discovery and materials science.
Conclusion
Successful molecular property prediction requires a holistic approach that integrates thoughtful data preparation, appropriate model selection, rigorous validation, and a clear understanding of uncertainty. Foundational choices in molecular representation and dataset understanding directly impact model performance, while advanced methodologies like generative models and automated feature selection open new avenues for molecular design. Crucially, practitioners must prioritize troubleshooting for dataset bias and activity cliffs, and employ robust, statistically sound validation practices to avoid over-optimism. As the field advances, the integration of more sophisticated uncertainty quantification and a focus on clinically relevant evaluation metrics will be paramount. By adhering to these principles, machine learning can transition from a promising tool to a reliable component in the drug discovery pipeline, ultimately accelerating the development of new therapeutics and enhancing confidence in computational predictions.
References
- 1. Machine Learning for Drug Development - Zitnik Lab [https://zitniklab.hms.harvard.edu/drugml/]
- 2. Molecular property prediction in the ultra‐low data regime [https://www.nature.com/articles/s42004-025-01592-1]
- 3. BOOM: Benchmarking Out-Of-distribution Molecular ... [https://arxiv.org/abs/2505.01912]
- 4. Geometric deep learning for molecular property predictions ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00895-0]
- 5. New machine-learning application to help researchers ... [https://news.mit.edu/2025/chemxploreml-app-helps-predict-chemical-properties-0724]
- 6. PatWalters/resources_2025: Machine Learning in Drug ... [https://github.com/PatWalters/resources_2025]
- 7. Advanced Machine Learning and Molecular Simulations for ... [https://www.schrodinger.com/materials-science/learn/white-paper/advanced-machine-learning-and-molecular-simulations-for-formulation-design/]
- 8. Could graph neural networks learn better molecular ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-020-00479-8]
- 9. Graph neural networks for molecular and materials ... [https://www.oaepublish.com/articles/jmi.2023.10]
- 10. Open Molecular Crystals 2025 (OMC25) Dataset and Models [https://ai.meta.com/research/publications/open-molecular-crystals-2025-omc25-dataset-and-models/]
- 11. [2505.08762] The Open Molecules 2025 (OMol25) Dataset, ... [https://arxiv.org/abs/2505.08762]
- 12. A Record-Breaking Dataset to Train AI Models has Launched [https://newscenter.lbl.gov/2025/05/14/computational-chemistry-unlocked-a-record-breaking-dataset-to-train-ai-models-has-launched/]
- 13. Kolmogorov–Arnold graph neural networks for molecular ... [https://www.nature.com/articles/s42256-025-01087-7]
- 14. FGBench: A Dataset and Benchmark for Molecular ... [https://arxiv.org/abs/2508.01055]
- 15. Understanding the Capabilities of Molecular Graph Neural ... [https://arxiv.org/html/2505.12137v1]
- 16. Meta OMol25 Electronic Structures Dataset | MDF Spotlight [https://www.materialsdatafacility.org/spotlight/omol25]
- 17. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 18. Out-of-Distribution Property Prediction in Materials and ... [https://www.nature.com/articles/s41524-025-01808-x]
- 19. FGBench: A Dataset and Benchmark for Molecular ... [https://arxiv.org/html/2508.01055v1]
- 20. Enhancing molecular property prediction through data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12574261/]
- 21. Comparative Evaluation of Applicability Domain Definition ... [https://arxiv.org/html/2411.00920v1]
- 22. Comparison of Different Approaches to Define the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6268288/]
- 23. A general approach for determining applicability domain of ... [https://www.nature.com/articles/s41524-025-01573-x]
- 24. Applicability domains are common in QSAR but irrelevant ... [https://variational.ai/blog/applicability-domains-are-common-in-qsar-but-irrelevant-for-conventional-ml-tasks/]
- 25. Applicability domain-expansion studies for machine ... [https://pubmed.ncbi.nlm.nih.gov/41061300/]
- 26. Applicability Domain Expansion Studies for Machine ... [https://www.sciencedirect.com/science/article/abs/pii/S009095562509169X]
- 27. Impact of Applicability Domains to Generative Artificial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10308412/]
- 28. Federated Learning for ADMET Prediction [https://www.apheris.com/resources/blog/federated-learning-for-admet-prediction-expanding-model-applicability]
- 29. Deterministic vs. Probabilistic Deep Learning [https://towardsdatascience.com/deterministic-vs-probabilistic-deep-learning-5325769dc758/]
- 30. Deterministic vs. probabilistic models: Guide for data teams [https://www.rudderstack.com/blog/deterministic-vs-probabilistic/]
- 31. Multi-task methods for molecular property prediction [https://www.sciencedirect.com/science/article/pii/S0098135425002571]
- 32. Upgrading Reliability in Molecular Property Prediction by ... [https://pubmed.ncbi.nlm.nih.gov/41060699/]
- 33. Understanding the Three Faces of AI: Deterministic ... [https://www.mymobilelyfe.com/artificial-intelligence/understanding-the-three-faces-of-ai-deterministic-probabilistic-and-generative/]
- 34. Probabilistic Machine Learning: An Introduction [https://probml.github.io/pml-book/book1.html]
- 35. Applications of machine learning in drug discovery and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6552674/]
- 36. Crash Course on Machine Learning for Molecular Design [https://roinaveiro.github.io/ml4md-course/]
- 37. Advancing Molecular Property Prediction for Environmental ... [https://www.sciencedirect.com/science/article/abs/pii/S0169743925002667]
- 38. Unified and explainable molecular representation learning ... [https://www.nature.com/articles/s41467-025-63730-6]
- 39. Molecular Graph Representation Learning via Structural ... [https://arxiv.org/abs/2409.08580]
- 40. A Transformer-based Generative Model for De Novo ... [https://arxiv.org/abs/2210.08749]
- 41. De novo molecular design and generative models [https://pubmed.ncbi.nlm.nih.gov/34082136/]
- 42. A Comparative Analysis of Cheminformatics Platforms [https://intuitionlabs.ai/articles/cheminformatics-platforms-comparison]
- 43. Machine learning and feature selection for drug response ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6381361/]
- 44. Comparative evaluation of feature reduction methods for ... [https://www.nature.com/articles/s41598-024-81866-1]
- 45. Automatic feature selection and weighting in molecular ... [https://www.nature.com/articles/s41467-024-55449-7]
- 46. Automatic feature selection and weighting in molecular ... [https://arxiv.org/abs/2411.00851]
- 47. Automatic feature selection and weighting in molecular ... [https://www.semanticscholar.org/paper/b1a0625693c8a0291b16b2ff2ed3122a925fd112]
- 48. Machine Learning Methods for Small Data Challenges in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10999174/]
- 49. Small data machine learning in materials science [https://www.nature.com/articles/s41524-023-01000-z]
- 50. Bias recognition and mitigation strategies in artificial ... [https://www.nature.com/articles/s41746-025-01503-7]
- 51. A Framework for Identifying, Assessing and Mitigating ... [https://virtualcellmodels.cziscience.com/micro-pub/bias-assessment-guide]
- 52. Uncertainty-biased molecular dynamics for learning ... [https://www.nature.com/articles/s41524-024-01254-1]
- 53. Interpretability With Accurate Small Models [https://www.frontiersin.org/journals/artificial-intelligence/articles/10.3389/frai.2020.00003/full]
- 54. Accelerating AI Drug Discovery with Open Source Datasets [https://www.recursion.com/news/accelerating-ai-drug-discovery-with-open-source-datasets]
- 55. A scoping review of self-supervised representation ... [https://www.nature.com/articles/s41746-025-01692-1]
- 56. Deep Learning vs Machine Learning: Key Differences [https://labelyourdata.com/articles/deep-learning-vs-machine-learning]
- 57. Representation Potentials of Foundation Models for ... [https://arxiv.org/html/2510.05184v1]
- 58. Advanced machine learning for innovative drug discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12333061/]
- 59. Explainable uncertainty quantifications for deep learning ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00682-3]
- 60. Uncertainty quantification for neural network potential ... [https://www.nature.com/articles/s41524-025-01572-y]
- 61. Uncertainty Color Palettes | Data Visualization Blog [https://sathvikpal.github.io/Data_Visualization_Studio/readings/2018/12/01/uncertainty.html]
- 62. Best Color Palettes for Scientific Figures and Data ... [https://www.simplifiedsciencepublishing.com/resources/best-color-palettes-for-scientific-figures-and-data-visualizations]
- 63. Uncertainty Calibration in Molecular Machine Learning [https://chemrxiv.org/engage/chemrxiv/article-details/68f1fb38bc2ac3a0e0805b98]
- 64. Analysis in Uncertainty Using... Predicting Molecular Properties [https://research.ibm.com/publications/uncertainty-analysis-in-predicting-molecular-properties-using-chemical-foundation-models]
- 65. [2504.02606] Improving Counterfactual Truthfulness for Molecular ... [https://arxiv.org/abs/2504.02606]
- 66. Activity Cliff-Informed Contrastive Learning for Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11643338/]
- 67. Evolving Concept of Activity Cliffs - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC6740043/]
- 68. Prediction of activity cliffs on the basis of images using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8636405/]
- 69. Exploring QSAR models for activity-cliff prediction [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00708-w]
- 70. RL-GCL: Reinforcement Learning-Guided Contrastive ... [https://www.sciencedirect.com/science/article/abs/pii/S1566253525002817]
- 71. Reinforcement Learning for Enhanced Targeted Molecule ... [https://arxiv.org/html/2405.06836v2]
- 72. MoleculeNet: a benchmark for molecular machine learning [https://pmc.ncbi.nlm.nih.gov/articles/PMC5868307/]
- 73. A systematic study of key elements underlying molecular ... [https://www.nature.com/articles/s41467-023-41948-6]
- 74. MolFCL: predicting molecular properties through chemistry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11878793/]
- 75. MoleculeNet: a benchmark for molecular machine learning [https://pubs.rsc.org/en/content/articlelanding/2018/sc/c7sc02664a]
- 76. We Need Better Benchmarks for Machine Learning in Drug ... [http://practicalcheminformatics.blogspot.com/2023/08/we-need-better-benchmarks-for-machine.html]
- 77. A systematic study of key elements underlying molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10575948/]
- 78. Analyzing Learned Molecular Representations for Property ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6727618/]
- 79. Data splitting to avoid information leakage with DataSAIL [https://pmc.ncbi.nlm.nih.gov/articles/PMC11978981/]
- 80. Some Thoughts on Splitting Chemical Datasets [http://practicalcheminformatics.blogspot.com/2024/11/some-thoughts-on-splitting-chemical.html]
- 81. Dataset Splits - TDC [https://tdcommons.ai/functions/data_split/]
- 82. UMAP-based clustering split for rigorous evaluation of AI ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12153141/]
- 83. Splitting dataset with splito - Datamol.io [https://splito-docs.datamol.io/stable/tutorials/The_Basics.html]
- 84. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 85. The topology of molecular representations and its influence on ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01045-w]
- 86. Deep learning methods for molecular representation and ... [https://www.sciencedirect.com/science/article/pii/S135964462200366X]
- 87. Advancements in Molecular Property Prediction: A Survey ... [https://arxiv.org/html/2408.09461v1]
- 88. Evaluation metrics and statistical tests for machine learning [https://www.nature.com/articles/s41598-024-56706-x]
- 89. Evaluation Metrics in Machine Learning [https://www.geeksforgeeks.org/machine-learning/metrics-for-machine-learning-model/]
- 90. 12 Important Model Evaluation Metrics for Machine ... [https://www.analyticsvidhya.com/blog/2019/08/11-important-model-evaluation-error-metrics/]
- 91. Context-informed few-shot molecular property prediction via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12510055/]
- 92. Enhancing molecular property prediction through data ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01103-3]