Active Learning for Chemical Space Exploration: Strategies, Applications, and Future Directions in Drug Discovery
This article provides a comprehensive overview of active learning (AL) methodologies for navigating vast chemical spaces in drug discovery and materials science.
Active Learning for Chemical Space Exploration: Strategies, Applications, and Future Directions in Drug Discovery
Abstract
This article provides a comprehensive overview of active learning (AL) methodologies for navigating vast chemical spaces in drug discovery and materials science. It covers foundational concepts, including the challenge of exploring immense molecular libraries and the core AL iterative loop. The review details cutting-edge methodological strategies, from multi-level Bayesian optimization to alchemical free energy calculations and machine learning-guided docking. It addresses critical troubleshooting aspects for low-data regimes and model generalizability, and presents rigorous validation through case studies and performance benchmarks against traditional methods. Aimed at researchers and drug development professionals, this article synthesizes recent advances to guide the effective implementation of AL for accelerated molecular discovery.
Navigating the Vastness of Chemical Space: The Active Learning Imperative
The concept of "chemical space" encompasses all possible organic molecules and materials of interest for drug discovery, a universe estimated to contain up to 10^60 drug-like compounds [1]. This vast expanse presents both extraordinary opportunity and formidable challenge for researchers seeking new therapeutics. The size of actionable chemical spaces is surging due to novel computational and experimental techniques, generating novel molecular matter that cannot be neglected in early-phase drug discovery [2]. Huge, combinatorial, make-on-demand chemical spaces with high probability of synthetic success rise exponentially in content, generative machine learning models work alongside synthesis prediction, and DNA-encoded libraries offer new ways of hit structure discovery [2]. These technologies enable searching for new chemical matter in a much broader and deeper manner with less effort and fewer financial resources, yet they simultaneously create unprecedented cheminformatics challenges for making these spaces searchable and analyzable [2].
The 'Big Data' era in medicinal chemistry presents new challenges for analysis. While modern computers can store and process millions of molecular structures, final decisions in medicinal chemistry remain in human hands [3]. However, the ability of humans to analyze large chemical data sets is limited by cognitive constraints, creating a demand for methods and tools to visualize chemical space and facilitate navigation [3]. This whitepaper examines the current state of chemical space exploration, focusing specifically on the transformative role of active learning methodologies in bridging the gap between theoretical possibility and practical drug discovery.
Representing and Visualizing Chemical Space
Chemical Space Networks and Molecular Complexity
Traditional coordinate-based representations of chemical space face significant limitations, including lack of invariance to chosen features and difficulty handling both discrete and continuous features [4]. Chemical Space Networks (CSNs) have emerged as a powerful alternative, representing chemicals as nodes connected by edges based on molecular similarity [4]. This approach allows application of graph theory metrics—degree, betweenness, and eigenvector centrality—to characterize chemical behavior within the network. Research has demonstrated that CSNs exhibit complex non-random organization and can reveal meaningful structural patterns related to biological activity, such as in developmental toxicity prediction where CSNs highlight well-established toxicophores like aryl derivatives, neurotoxic hydantoins, barbiturates, and amino alcohols [4].
Molecular complexity represents another critical dimension in chemical space navigation, reflecting the diversity and intricacy of substructures in a molecule and indicating synthetic difficulty [5]. While medicinal chemists often navigate a "love-hate relationship" with complexity—appreciating simple, elegant molecules while recognizing the need for added complexity to optimize potency, selectivity, and metabolic stability—recent work has proposed simplified complexity measures [5].
Table 1: Molecular Complexity and Synthetic Accessibility Measures
| Name | Type | Description | Reference |
|---|---|---|---|
| MC1 | Complexity | Fraction of non-divalent nodes in molecular graph | [5] |
| MC2 | Complexity | Number of non-divalent nodes (excluding certain carbonyl groups) | [5] |
| FCFP4 | Complexity | Number of on-bits in a binary 2048-bit FCFP4 fingerprint | [5] |
| Data Warrior | Complexity | Fractal complexity using Minkowski–Bouligand dimension concept | [5] |
| SAscore | Synthesizability | Fragment occurrence combined with complexity penalty | [5] |
| SCS | Synthesizability | Machine-learned score predicting synthetic steps from Reaxys data | [5] |
Visualization Approaches for High-Dimensional Data
As chemical libraries grow to millions of compounds, efficient visualization methods become increasingly important [3]. Recent advances include tree-maps (TMAPs) organized by substructure similarity using molecular fingerprints like MAP4, where each molecule is a point color-coded according to properties or complexity metrics [5]. Deep generative modeling combined with chemical space visualization paves the way for interactive exploration of chemical space, extending beyond chemical compounds to include reactions and chemical libraries [3]. These visualization approaches also support visual validation of QSAR/QSPR models and analysis of activity/property landscapes [3].
Figure 1: Chemical Space Visualization Workflow
Active Learning for Chemical Space Exploration
The Active Learning Paradigm in Drug Discovery
Active learning represents a paradigm shift in computational drug discovery, addressing the fundamental limitation of traditional virtual screening: the inability to evaluate all compounds in ultralarge libraries [6]. The core concept involves an iterative approach where machine learning models suggest new compounds for evaluation by an oracle (experimental measurement or computational predictor), with these compounds and their scores incorporated back into the training set for continuous model improvement [1]. This methodology is particularly valuable in low-data scenarios typical of drug discovery, where it can achieve up to a sixfold improvement in hit discovery compared to traditional screening methods [6].
In practice, active learning driven prioritization has demonstrated significant utility in target-focused drug discovery. For example, in targeting the SARS-CoV-2 main protease (Mpro), researchers have interfaced structure-based growing algorithms with active learning to improve the efficiency of searching the combinatorial space of possible linkers and functional groups [7]. This approach successfully identified several small molecules with high similarity to molecules discovered by the COVID moonshot effort, using only structural information from a fragment screen in a fully automated fashion [7].
Active Learning Strategies and Configurations
Several strategic approaches exist for compound selection within active learning cycles, each with distinct advantages for different discovery scenarios:
- Greedy Selection: Chooses only the top predicted binders at every iteration step, focusing exclusively on exploitation of the current model [1].
- Uncertainty Selection: Selects ligands with the largest prediction uncertainty, prioritizing exploration of chemical space regions where the model is least confident [1].
- Mixed Strategy: Identifies top predicted binders then selects those with the most uncertain predictions, balancing exploration and exploitation [1].
- Narrowing Strategy: Combines broad selection in initial iterations with subsequent switch to greedy approach, beginning with exploration before focusing on exploitation [1].
Table 2: Active Learning Performance Across Molecular Representations
| Representation | Description | Best For | Limitations |
|---|---|---|---|
| 2D_3D Features | Constitutional, electrotopological, and molecular surface area descriptors | General-purpose screening | Computationally intensive |
| Atom-hot | Grid-based 3D voxel counting of ligand atoms | Structure-based design | Requires binding poses |
| PLEC Fingerprints | Protein-ligand interaction fingerprints | Target-specific optimization | Protein structure dependent |
| MDenerg | Residue-based interaction energies | Accurate binding affinity prediction | Computationally expensive |
| R-group-only | Focused on variable substituents | Lead optimization series | Limited scaffold diversity |
The effectiveness of active learning depends critically on both the molecular representation and the selection strategy. Studies have systematically analyzed active learning strategies combined with deep learning architectures on large-scale molecular libraries, identifying the most important determinants of success in low-data regimes [6]. Optimal performance typically requires matching the exploration strategy to the specific drug discovery stage—broad exploration for early hit identification versus focused exploitation for lead optimization.
Experimental Protocols and Case Studies
Integrated Active Learning Workflow for SARS-CoV-2 Mpro
A recent study demonstrated a complete active learning workflow for targeting SARS-CoV-2 main protease (Mpro) using the FEgrow software package [7]. The methodology provides a template for structure-based active learning applications:
Initialization Phase:
- Protein Preparation: Obtain and prepare the receptor structure from crystallographic data (PDB ID: 7BQY for Mpro)
- Ligand Core Definition: Define a constrained core based on fragment hits from crystallographic screens
- Chemical Space Definition: Specify linkers (from library of 2000) and R-groups (from library of ~500) or seed with purchasable compounds from on-demand libraries like Enamine REAL
Active Learning Cycle:
- Compound Generation: FEgrow builds ligands by growing user-defined functional groups off the constrained core
- Pose Optimization: Hybrid machine learning/molecular mechanics (ML/MM) potential energy functions optimize bioactive conformers
- Scoring: gnina convolutional neural network scoring function predicts binding affinity
- Model Training: Random forest or neural network models trained on scored compounds
- Compound Selection: Mixed strategy selection of additional compounds for next cycle
- Iteration: Repeated for 10-20 cycles or until convergence
Experimental Validation:
- Compound Procurement: Top-ranked compounds ordered from on-demand libraries
- Activity Testing: Fluorescence-based Mpro assay to confirm inhibitory activity
- Hit Confirmation: Dose-response measurements for active compounds
This protocol successfully identified three weakly active Mpro inhibitors from 19 designed compounds, demonstrating the feasibility of fully automated structure-based design guided by active learning [7].
Figure 2: Active Learning Cycle for Drug Discovery
Prospective PDE2 Inhibitor Discovery with Alchemical Free Energy Calculations
Another sophisticated implementation combined active learning with alchemical free energy calculations for phosphodiesterase 2 (PDE2) inhibitor discovery [1]. This approach addresses the critical need for accurate binding affinity prediction in chemical space navigation:
Ligand Pose Generation Protocol:
- Reference Selection: Identify reference crystal structures (4D08, 4D09, 4HTX for PDE2)
- Similarity Matching: For each library compound, select reference with highest Dice similarity based on RDKit topological fingerprint
- Constrained Embedding: Generate initial poses using ETKDG algorithm with core atoms constrained to reference geometry
- Pose Refinement: Molecular dynamics simulation in vacuum with hybrid topology morphing
Active Learning with Free Energy Oracle:
- Initialization: Weighted random selection based on t-SNE embedding diversity
- Free Energy Calculations: Relative binding free energies using alchemical transformation
- Model Training: Gaussian process regression or neural networks on free energy data
- Batch Selection: 100 compounds per iteration using mixed strategy
- Termination: After 10-15 iterations or when high-affinity binders identified
This prospective application successfully identified potent PDE2 inhibitors while explicitly evaluating only a small fraction (∼5%) of the virtual library, demonstrating the efficiency gains possible through active learning [1].
Emerging Frontiers and Research Directions
Chemical-Linguistic Space Integration
An emerging frontier involves navigating chemical-linguistic sharing space through heterogeneous molecular encoding [8]. This approach addresses the semantic gap between natural language and molecular representations by unifying features from multiple perspectives:
- 1D SMILES Encoder: Embeds SMILES strings into text feature space
- 2D Graph Encoder: Captures molecular connectivity and topology
- 3D Coordinate Encoder: Incorporates spatial atomic arrangements
- Molecular Fragment Encoder: Provides explicit substructure information through learned fragments
The Heterogeneous Molecular Encoding (HME) framework compresses molecular features from fragment sequence, topology, and conformation with Q-learning, enabling more effective bidirectional mapping between chemical structures and textual descriptions [8]. This facilitates both chemical space exploration with linguistic guidance and linguistic space exploration with molecular guidance.
Multi-Objective Optimization
Future directions increasingly focus on multi-objective optimization, simultaneously balancing potency, selectivity, synthetic accessibility, and pharmacokinetic properties [6]. Active learning protocols are being extended to incorporate:
- Synthetic Complexity Scores (SCS): Machine-learned predictions of synthetic steps from Reaxys data [5]
- Molecular Complexity Metrics: MC1 and MC2 for synthetic accessibility assessment [5]
- Property Predictions: Integrated models for ADMET and physicochemical properties
- Multi-fidelity Approaches: Combining cheap approximate predictions with expensive accurate calculations
Table 3: Key Research Reagents and Computational Tools for Chemical Space Exploration
| Tool/Resource | Type | Function | Access |
|---|---|---|---|
| FEgrow | Software | Builds and scores congeneric ligand series in protein binding pockets | Open-source [7] |
| Enamine REAL | Compound Library | >5.5 billion make-on-demand compounds for virtual screening | Commercial [7] |
| RDKit | Cheminformatics | Core molecular informatics functionality for fingerprinting and similarity | Open-source [7] |
| OpenMM | Molecular Dynamics | Platform for molecular simulation and energy minimization | Open-source [7] |
| gnina | Docking Software | Convolutional neural network scoring for protein-ligand binding | Open-source [7] |
| GDB Databases | Chemical Space | Enumerated hypothetical molecules from mathematical graphs | Research use [5] |
| TMAP | Visualization | Tree-based visualization of high-dimensional chemical space | Open-source [5] |
| PLIP | Analysis | Protein-ligand interaction profiling for structural motifs | Open-source [7] |
The grand challenge of navigating 10^60 possible compounds to practical drug discovery is being transformed by active learning approaches. By combining sophisticated molecular representations with iterative experimental design, researchers can now efficiently explore relevant regions of chemical space that would otherwise remain inaccessible. The integration of structure-based design, synthetic accessibility metrics, and multi-objective optimization within active learning frameworks represents a paradigm shift in early-phase drug discovery. As these methodologies continue to mature, incorporating emerging capabilities in chemical-linguistic space navigation and heterogeneous molecular encoding, they promise to significantly accelerate the identification of novel therapeutic candidates while reducing resource requirements. The future of chemical space exploration lies not in exhaustive enumeration, but in intelligent navigation—a journey guided by active learning from theoretical possibility to practical discovery.
Active learning represents a paradigm shift in machine learning for scientific discovery, moving from passive model training on static datasets to an intelligent, iterative process that optimizes data acquisition. In the context of chemical space exploration—a domain characterized by vast molecular diversity and computationally expensive data generation—active learning has emerged as a critical strategy for accelerating research. This approach strategically selects the most informative data points for labeling and model training, dramatically reducing computational costs while maintaining or even improving model accuracy. For researchers and drug development professionals, implementing active learning iterative loops enables efficient navigation of complex chemical landscapes that would otherwise be prohibitively expensive to explore exhaustively. The core principle hinges on recognizing that not all data points contribute equally to model improvement, and that selectively querying the most valuable samples creates a self-improving cycle that maximizes learning efficiency while minimizing resource expenditure [9] [10].
The Active Learning Iterative Loop: Core Components and Workflow
Fundamental Operating Principle
The active learning iterative loop operates through a cyclic process of model training, data selection, and knowledge incorporation. Unlike traditional supervised learning that uses a fixed, pre-defined dataset, active learning algorithms actively query a human expert or information source to annotate specifically chosen data points. The primary objective is to minimize the labeled data required for training while maximizing model performance, creating an efficient learning trajectory that focuses resources on the most chemically relevant regions of feature space. This approach is particularly valuable in chemical research where obtaining labeled data through experimentation or simulation is costly, time-consuming, or scarce [9].
The loop begins with a small initial set of labeled data points, which serves as the starting point for training the first model iteration. Through successive cycles, the model identifies gaps in its knowledge and strategically requests annotations for samples that will provide the greatest information gain. This creates a continuous improvement mechanism where each iteration enhances the model's understanding of chemical space, enabling more informed exploration in subsequent cycles [9] [10].
The Five-Step Iterative Process
The active learning framework implements a structured, five-step process that transforms raw data into refined predictive models:
Step 1: Planning and Requirements - Researchers define project objectives and outline essential requirements that must be met for project success. This establishes the foundation for all subsequent iterations and ensures the active learning process remains aligned with research goals [11].
Step 2: Analysis and Design - The team focuses on the specific business needs and technical requirements of the project. This phase involves designing the initial model architecture and determining which chemical features or properties will be prioritized during the exploration process [11].
Step 3: Implementation - The development team creates the first iteration of the active learning model. This initial implementation aims to achieve the primary project objectives while establishing the framework for subsequent refinement cycles [11].
Step 4: Testing - The current model iteration undergoes rigorous evaluation, typically through validation against known chemical data or through computational experiments. In chemical space exploration, this often involves comparing predictions against established quantum mechanical calculations or experimental results [10] [11].
Step 5: Evaluation and Review - Researchers assess the iteration's success and identify necessary improvements. The model's performance is analyzed to determine which chemical regions require additional sampling. Based on this evaluation, the process returns to Step 2 to create the next iteration, with each cycle building upon previous knowledge while maintaining focus on the original project objectives [11].
This iterative methodology stands in contrast to non-iterative approaches like the Waterfall method, where project phases are sequentially completed without revisiting previous stages. The iterative nature of active learning provides the flexibility needed to adapt to new insights discovered during chemical space exploration [11].
Workflow Visualization
The following diagram illustrates the core active learning iterative loop as implemented in chemical research:
Figure 1: Active Learning Iterative Loop
Key Query Strategies for Chemical Space Exploration
Query strategies form the decision-making engine of the active learning loop, determining which unlabeled samples would provide maximum information gain if annotated. In chemical space exploration, these strategies guide the efficient sampling of molecular configurations, reactions, or properties. Three primary configurations are commonly implemented in research settings:
Pool-Based Sampling - The model assesses all available unlabeled data points in a "pool" and queries the most informative samples based on specific criteria. This approach is particularly effective when researchers have access to large databases of uncharacterized molecules or molecular configurations [12].
Stream-Based Selective Sampling - Each data point is individually considered as it becomes available, with the model determining whether to query it or reject it based on current information needs. This method is valuable when chemical data is generated continuously, such as in real-time monitoring of reactions or high-throughput computational screening [9].
Membership Query Synthesis - The model generates its own synthetic samples for annotation, creating novel molecular structures or configurations not present in the original dataset. This advanced approach enables exploration beyond known chemical spaces but requires careful validation to ensure synthetic samples remain chemically plausible [12].
Specific Query Algorithms
Within these broader configurations, several algorithmic approaches determine how "informativeness" is quantified for sample selection:
Table 1: Active Learning Query Strategies
| Strategy | Mechanism | Chemical Application Context |
|---|---|---|
| Random Sampling | Selects samples randomly from unlabeled pool | Establishing baseline performance; initial diverse sampling |
| Least Confidence Sampling | Prioritizes samples with lowest prediction confidence | Identifying regions of chemical space with high uncertainty |
| Entropy Sampling | Selects samples with highest entropy in prediction distribution | Molecular property prediction where multiple outcomes are plausible |
| Discriminative Active Learning | Chooses samples to make labeled and unlabeled sets indistinguishable | Ensuring representative coverage of diverse chemical classes |
Each strategy offers distinct advantages for different phases of chemical space exploration. Least Confidence and Entropy Sampling are particularly valuable for identifying regions where quantum mechanical calculations would provide maximum information gain, while Discriminative approaches ensure comprehensive coverage of chemical diversity [12].
Implementation Framework: PALIRS for Infrared Spectra Prediction
The practical implementation of active learning for chemical space exploration is exemplified by PALIRS (Python-based Active Learning Code for Infrared Spectroscopy), a recently developed framework for efficiently predicting IR spectra of organic molecules. This system addresses a critical challenge in computational chemistry: the accurate simulation of IR spectra traditionally requires computationally expensive density functional theory-based ab-initio molecular dynamics (AIMD) calculations. PALIRS overcomes this limitation by implementing an active learning-enhanced machine-learned interatomic potential (MLIP) that achieves accurate spectral predictions at a fraction of the computational cost [10].
The PALIRS framework demonstrates how active learning iterative loops can be specifically designed for chemical applications. By focusing sampling efforts on molecular configurations that maximize information gain, the system efficiently explores the relevant chemical space while minimizing quantum mechanical computations. This approach has shown particular success for small catalytically relevant organic molecules, accurately reproducing IR spectra computed with AIMD while dramatically reducing computational requirements [10].
Detailed Workflow and Experimental Protocol
The PALIRS implementation follows a structured four-step methodology for IR spectrum prediction:
Step 1: Active Learning for MLIP Development - Initial machine-learned interatomic potentials are trained on molecular geometries sampled along normal vibrational modes. An active learning strategy then iteratively expands the training set through machine learning-assisted molecular dynamics (MLMD) simulations. The acquisition function selects molecular configurations with the highest uncertainty in force predictions, enriching the dataset with the most informative structures while minimizing redundancy. To balance exploration and exploitation, MLMD simulations are performed at multiple temperatures (300K, 500K, and 700K) [10].
Step 2: Dipole Moment Model Training - A separate machine learning model, based on the MACE framework, is specifically trained to predict dipole moments necessary for IR spectra calculations. This specialization ensures accurate representation of the electronic properties critical for spectral simulations [10].
Step 3: MLMD Production Simulations - Using the refined MLIP for energies and forces, researchers conduct production molecular dynamics simulations. The trajectory provides the structural evolution data required for spectral calculation [10].
Step 4: IR Spectrum Calculation - Dipole moments are computed for all structures along the trajectory using the specialized dipole model. IR spectra are then derived by computing the autocorrelation function of these dipole moments, reproducing the anharmonic effects captured by more computationally intensive methods [10].
The iterative active learning process in PALIRS continues until model performance plateaus or computational budgets are exhausted. Through 40 active learning iterations, the final dataset consists of 16,067 structures (approximately 600-800 structures per molecule), significantly fewer than would be required for exhaustive sampling [10].
Workflow Visualization
The following diagram details the PALIRS active learning workflow for infrared spectra prediction:
Figure 2: PALIRS Active Learning Workflow
Research Reagents and Computational Tools
Successful implementation of active learning for chemical space exploration requires specific computational tools and methodological components:
Table 2: Essential Research Reagents and Computational Tools
| Component | Function | Implementation Example |
|---|---|---|
| Initial Dataset | Provides starting point for active learning loop | Molecular geometries from normal mode sampling [10] |
| MLIP Architecture | Machine-learned interatomic potential for energy/force prediction | MACE (Multi-Atomic Cluster Expansion) models [10] |
| Uncertainty Quantification | Estimates model confidence for sample selection | Ensemble of three MACE models [10] |
| Query Strategy | Selects most informative samples for annotation | Highest uncertainty in force predictions [10] |
| Ab-Initio Calculator | Provides ground-truth labels for selected samples | FHI-aims DFT code [10] |
| Active Learning Framework | Manages iterative learning process | PALIRS (Python-based Active Learning Code) [10] |
Performance Assessment and Validation
Quantitative Performance Metrics
The effectiveness of active learning iterative loops must be rigorously quantified through appropriate performance metrics. In the PALIRS implementation, researchers evaluated model improvement during active learning by comparing predictions against a predefined test set of harmonic frequencies. These metrics provide reliable validation of the model's accuracy and progression through successive iterations [10].
Key quantitative assessments include:
Mean Absolute Error (MAE) - Measures the average magnitude of errors between MLIP-computed harmonic frequencies and DFT reference values, providing a comprehensive view of model accuracy across the chemical space of interest [10].
Learning Curves - Track model performance as a function of training set size, demonstrating the efficiency gains achieved through strategic sample selection compared to random sampling approaches [10].
Computational Cost Reduction - Quantifies the reduction in required quantum mechanical calculations while maintaining target accuracy levels. The PALIRS framework demonstrated accurate IR spectrum prediction at a fraction of the computational cost of traditional AIMD approaches [10].
Advantages and Limitations
The implementation of active learning iterative loops offers significant advantages for chemical space exploration, along with some important considerations:
Advantages:
- Reduced Labeling Costs - By selectively choosing the most informative samples for expensive quantum mechanical calculations, active learning significantly reduces computational requirements [9] [10].
- Improved Accuracy - Focusing on high-information regions of chemical space often produces more accurate models than training on larger but less informative datasets [9].
- Faster Convergence - Strategic sample selection enables models to achieve target performance levels with fewer training iterations [9].
- Adaptability - The iterative nature allows models to adapt to new discoveries and refine their understanding of chemical space throughout the exploration process [9].
Challenges and Limitations:
- Initial Data Requirements - Effective active learning requires sufficient initial data to bootstrap the iterative process, which can be challenging for novel chemical domains [10].
- Uncertainty Quantification - Accurate estimation of model uncertainty is critical for sample selection but can be computationally expensive or methodologically complex [10].
- Temporal Overhead - The iterative cycle of training, selection, and annotation introduces organizational complexity compared to single-batch training [11].
- Potential for Bias - If not properly managed, active learning can over-exploit certain regions of chemical space while neglecting others [9].
Future Directions and Integration with Foundation Models
The field of active learning for chemical space exploration is rapidly evolving, with several emerging trends shaping its future development. Recent advances in molecular foundation models like MIST (Molecular Insight SMILES Transformers) present opportunities for enhancing active learning frameworks. These models, trained on billions of molecular structures using novel tokenization schemes that capture nuclear, electronic, and geometric features, provide rich pretrained representations that can accelerate active learning iterations [13].
The integration of active learning with foundation models creates a powerful synergy: foundation models provide comprehensive initial representations of chemical space, while active learning efficiently targets computational resources to refine these representations for specific chemical properties or reactions. This combination is particularly valuable for exploring underrepresented regions of chemical space or extending models to novel molecular classes not well-represented in pretraining datasets [13].
Future methodological developments will likely focus on improving uncertainty quantification for complex molecular representations, developing multi-objective acquisition functions that balance multiple chemical priorities simultaneously, and creating more efficient integration between active learning cycles and high-throughput computational screening platforms. As these technical advances mature, active learning iterative loops will become increasingly central to computational chemical research, enabling more efficient exploration of the vast chemical universe and accelerating the discovery of novel molecules with valuable properties [10] [13].
In the pursuit of novel chemical entities, such as drugs or catalysts, researchers face a search space of astronomical proportions, estimated to contain up to 10^60 drug-like compounds [1]. Active learning (AL) has emerged as a powerful artificial intelligence (AI) paradigm to navigate this vast chemical space efficiently. An AL cycle operates as an iterative feedback loop: a machine learning (ML) model selects promising candidate molecules, an oracle evaluates them, and the results are used to retrain and improve the model for the next cycle [14]. While the ML model is often the focus of development, the nature of the oracle—the source of feedback—is equally critical for the success of any AL campaign.
The oracle provides the ground-truth data that guides the entire exploration process. It can be computational, using physics-based simulations to predict molecular properties, or experimental, relying on high-throughput laboratory measurements. The choice of oracle involves a fundamental trade-off between cost, throughput, and accuracy. This paper provides an in-depth examination of oracle definitions within AL for chemical space exploration, detailing their implementation, relative merits, and integration into robust experimental protocols for the drug development community.
Oracle Typology: Computational and Experimental Feedback
Oracles in active learning can be broadly categorized into two classes: those based on computational simulations and those relying on experimental data. The table below summarizes the primary oracle types used in the field.
Table 1: Types of Oracles Used in Active Learning for Chemical Discovery
| Oracle Type | Specific Method | Primary Output | Typical Use Case | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Computational | Alchemical Free Energy Calculations [15] [1] | Binding Affinity (ΔG) | Lead Optimization | High accuracy close to experiments; rigorous physical basis [1] | Computationally very demanding |
| Computational | Molecular Docking & Scoring (e.g., gnina) [7] | Docking Score | Virtual Screening & Hit Identification | Very high throughput; low cost per compound [7] | Approximate; can suffer from scoring errors |
| Computational | Hybrid ML/MM & Graph Neural Networks [16] [7] | Binding Affinity, Energy, Forces | Binding Pose Optimization & Property Prediction | Balances accuracy and speed; can enhance scoring [7] | Dependent on quality of training data |
| Experimental | High-Throughput In Vitro Assays [17] [14] | Inhibitory Activity (e.g., IC50), Toxicity | Experimental Validation & Toxicity Prediction | Provides direct, biological relevant data [17] | Costly and time-consuming relative to computation |
| Experimental | Fluorescence-Based Bioassay [7] | Enzyme Inhibition Activity | Confirmatory Testing of Designed Compounds | Direct functional readout | Lower throughput; requires compound synthesis |
Computational Oracles
Computational oracles provide a cost-effective way to evaluate vast regions of chemical space without the need for physical materials or complex laboratory setups.
Alchemical Free Energy Calculations: These methods are considered a high-accuracy computational oracle for predicting binding affinities. They work by using molecular dynamics simulations to compute the free energy difference between a ligand and a reference compound through a non-physical (alchemical) pathway [1]. While their accuracy is high, they are computationally demanding, often taking days to screen hundreds to thousands of ligands on high-performance computing clusters [1]. Consequently, they are ideally suited for the lead optimization phase, where precision is paramount, and are used as the oracle in AL cycles to train more efficient ML models [15] [1].
Molecular Docking and Scoring: In contrast, docking serves as a high-throughput computational oracle. Tools like gnina use convolutional neural networks to score how well a small molecule (ligand) fits into a protein's binding pocket [7]. This method is a cornerstone of virtual screening, allowing researchers to quickly triage millions of compounds. However, its approximations can lead to false positives and negatives. For instance, in a study targeting the SARS-CoV-2 main protease, the docking score from gnina was used as the primary oracle in an AL-driven workflow to prioritize compounds for synthesis [7].
Hybrid Machine Learning/Molecular Mechanics (ML/MM): Emerging approaches seek to balance speed and accuracy. The FEgrow software, for example, uses ML/MM potential energy functions to optimize the conformations of growing ligands within a protein binding pocket, using a docking score or other functions as its objective [7]. This creates a more refined, structure-based oracle for de novo molecular design.
Experimental Oracles
Experimental oracles provide the ultimate validation, as they measure real-world biological activity or properties.
High-Throughput In Vitro Assays: These are the workhorses of experimental feedback. Data from programs like the U.S. EPA ToxCast provide large-scale biological activity data that can be used to train and validate models for toxicity prediction [17]. In an AL context, such assays can be used directly as the oracle to select compounds for subsequent testing rounds, efficiently focusing experimental resources on the most informative candidates [14].
Confirmatory Biochemical Assays: In prospective drug discovery campaigns, computationally prioritized compounds are synthesized and tested in specific biochemical assays. For example, in the SARS-CoV-2 Mpro study, 19 designed compounds were ordered and tested in a fluorescence-based Mpro activity assay to confirm inhibitory activity, with three showing weak activity [7]. This experimental result closes the AL loop and provides hard validation of the overall strategy.
Implementation: Protocols and Workflows
Implementing an effective active learning system requires a carefully designed protocol that integrates the oracle, the machine learning model, and the chemical library.
General Active Learning Workflow
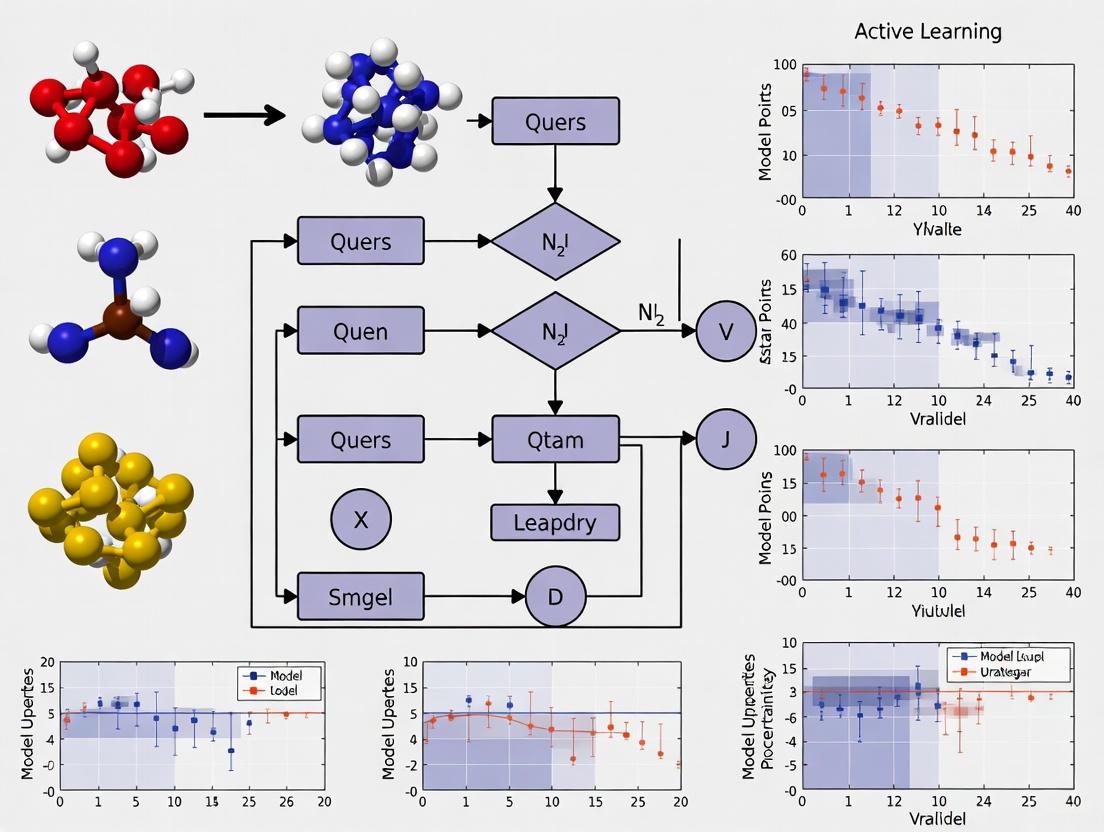
The following diagram illustrates the standard iterative cycle of an active learning campaign in chemical discovery.
This workflow is agnostic to the specific oracle used. The key steps are:
- Initialization: The process begins with a small, initially labeled set of compounds. This can be a random sample or a strategically chosen set to maximize diversity [1].
- Model Training: A machine learning model is trained on the current set of labeled data (e.g., compound structures and their associated oracle outputs).
- Prediction & Selection: The trained model predicts the properties of a large, unlabeled pool of compounds. A query strategy is then used to select the most promising candidates for evaluation. Common strategies include:
- Uncertainty Sampling: Selects compounds where the model's prediction is most uncertain, aiming to improve the model itself [7] [1].
- Greedy Selection: Selects the top-ranked compounds predicted to have the best properties (e.g., highest binding affinity), focusing on optimization [1].
- Mixed Strategy: Selects the best-predicted compounds from a subset where uncertainty is high, balancing exploration and exploitation [1].
- Oracle Evaluation: The selected candidates are evaluated by the chosen oracle, be it a free energy calculation, a docking run, or a biological experiment.
- Iteration: The newly acquired data is added to the training set, and the cycle repeats until a stopping criterion is met, such as a performance target or exhaustion of resources.
Case Study Protocol: AL with Free Energy Oracle
A robust protocol for using alchemical free energy calculations as an oracle was detailed by Khalak et al. [1]. The methodology below can be adapted for other oracles with appropriate modifications.
Table 2: Essential Research Reagents and Tools for an AL Cycle
| Category | Item / Software | Specification / Version | Critical Function in the Workflow |
|---|---|---|---|
| Chemical Library | Enamine REAL / In-house Library | >5.5 Billion Compounds [7] | Source of unlabeled candidate molecules for exploration. |
| Structure Preparation | RDKit [7] [1] | v.2020.09 or later | Canonicalization of SMILES, 2D/3D descriptor calculation, and fingerprint generation. |
| Binding Pose Generation | Hybrid Topology (pmx) [1] | N/A | Generates physically plausible ligand binding poses for free energy calculations. |
| Oracle Software | Gromacs [1] | 2021.1 or later | Performs molecular dynamics and alchemical free energy calculations. |
| Machine Learning | Scikit-learn, PyTorch, etc. | N/A | Builds models to predict oracle outcomes from molecular representations. |
| Representation | 2D_3D Features, PLEC Fingerprints [1] | N/A | Encodes molecular structure and protein-ligand interactions for ML input. |
Step-by-Step Protocol:
Library Curation and Preparation:
Ligand Pose Generation for a Structural Oracle:
- For each ligand, identify a reference crystal structure with a high-similarity inhibitor.
- Align the largest common substructure of the ligand to the reference inhibitor in the protein binding pocket.
- Use a constrained embedding algorithm (e.g., ETKDG in RDKit) to generate initial poses for the variable parts of the ligand [1].
- Refine the poses using molecular dynamics with strong restraints on the common core, "morphing" the reference inhibitor into the new ligand to ensure a physically realistic binding mode [1].
Feature Engineering for ML:
- Calculate fixed-size vector representations for each ligand. A comprehensive ("2D_3D") representation may include:
- For R-group optimization, consider using representations that focus only on the variable parts of the molecule to reduce noise.
Active Learning Cycle Execution:
- Initialization: Select an initial training set of 50-100 compounds using a diversity-oriented strategy, such as weighted random selection based on t-SNE clustering of molecular fingerprints [1].
- Oracle Evaluation: Run alchemical free energy calculations on the selected compounds to obtain binding affinities. This involves setting up hybrid topology systems and running thermodynamic integration (TI) or free energy perturbation (FEP) simulations [1].
- Model Training and Selection: Train multiple ML models (e.g., random forest, neural networks) using different molecular representations. Select the top-performing models based on cross-validation error.
- Candidate Selection: Apply the chosen query strategy (e.g., "mixed strategy") to the pooled library. The ML model predicts affinities and uncertainties for all unlabeled compounds. Select the next batch (e.g., 100 compounds) for oracle evaluation.
- Iteration: Continue the cycle for a predefined number of iterations (e.g., 10-20) or until a performance plateau is observed.
Performance and Validation
The effectiveness of an AL framework is ultimately judged by its ability to efficiently identify hits. Quantitative metrics from recent studies demonstrate this success.
Table 3: Performance Metrics of Active Learning Frameworks with Different Oracles
| Study & Application | Oracle Used | Key Performance Metric | Result | Data Efficiency |
|---|---|---|---|---|
| Thyroid Toxicity Prediction [17] | In Vitro Assay (ToxCast) | Matthews Correlation Coefficient (MCC) | 0.51 | Achieved with up to 73.3% less labeled data |
| SARS-CoV-2 Mpro Inhibitor Design [7] | Docking Score (gnina) & Fluorescence Assay | Experimental Hit Rate | 3 out of 19 tested compounds showed activity | Enabled prioritization from billions of compounds |
| PDE2 Inhibitor Discovery [1] | Alchemical Free Energy | Enrichment of Potent Binders | Identified high-affinity inhibitors | Required evaluation of only a small subset of the library |
The integration of computational and experimental feedback is powerfully exemplified in the work by Cree et al. [7]. Their AL-driven workflow for targeting SARS-CoV-2 Mpro used the gnina docking score as the primary computational oracle to prioritize compounds from the Enamine REAL library. The most promising designs were then synthesized and tested in a fluorescence-based biochemical assay, the experimental oracle. This closed loop allowed them to identify novel, active inhibitors based solely on initial fragment data, with several designs showing high similarity to those discovered by the large-scale COVID moonshot consortium [7]. This validates the entire AL pipeline, from computational pre-screening to experimental confirmation.
The "oracle" is the cornerstone of any active learning system for chemical discovery. Its definition—whether a computationally intensive free energy calculation, a high-throughput docking score, or a wet-lab assay—directly determines the cost, speed, and ultimate success of the exploration campaign. As the field advances, the integration of multiple oracles into a single workflow will become more prevalent, leveraging the speed of computational screens for broad exploration and the fidelity of experimental assays for final validation. Furthermore, the development of more accurate and efficient computational oracles, such as those based on hybrid ML/MM or advanced machine learning potentials trained on high-quality datasets like QDπ [18], will continue to narrow the gap between in silico prediction and experimental reality. By making informed choices about the oracle and meticulously implementing the associated protocols, researchers can dramatically accelerate the journey from a hypothesis to a novel, functional molecule.
The process of translating molecular structures into a computer-readable format, known as molecular representation, serves as the foundational step in computational chemistry and drug design [19]. It bridges the critical gap between chemical structures and their biological, chemical, or physical properties [19]. Effective molecular representation is paramount for various drug discovery tasks, including virtual screening, activity prediction, and scaffold hopping, as it enables researchers to navigate the vast chemical space efficiently and precisely [19]. The evolution of these methods has progressively enhanced our ability to characterize molecules, moving from simple, human-defined rules to complex, data-driven algorithms that capture deeper structural and functional relationships [19].
This evolution is particularly crucial within the framework of active learning for chemical space exploration. In this context, the choice of molecular representation directly influences how a model queries, selects, and prioritizes compounds for synthesis or testing [20]. The representation must not only encode chemical structure but also enable the model to efficiently explore regions of chemical space with desired biological properties, thereby accelerating the iterative cycle of prediction and experimental validation [19] [20].
Traditional Molecular Representation Methods
Traditional molecular representation methods rely on explicit, rule-based feature extraction. These methods are computationally efficient and have laid a strong foundation for many computational approaches in drug discovery [19].
Molecular Descriptors and Fingerprints
Molecular descriptors quantify the physical or chemical properties of molecules, such as molecular weight, hydrophobicity, or topological indices [19]. Molecular fingerprints, on the other hand, typically encode substructural information as binary strings or numerical values [19]. A prominent example is the Extended-Connectivity Fingerprint (ECFP), which represents local atomic environments in a compact and efficient manner, making it invaluable for representing complex molecules [19]. These representations are highly effective for tasks like similarity search, clustering, and quantitative structure-activity relationship (QSAR) modeling due to their interpretability and computational efficiency [19].
Table 1: Key Traditional Molecular Representation Methods
| Method Type | Specific Example | Representation Format | Primary Applications |
|---|---|---|---|
| Molecular Fingerprint | Extended-Connectivity Fingerprints (ECFP) | Binary or count-based vector | Similarity search, QSAR, clustering |
| Molecular Descriptor | alvaDesc descriptors | Numerical vector (e.g., molecular weight, logP) | QSAR/QSPR modeling, property prediction |
| String-Based | SMILES (Simplified Molecular-Input Line-Entry System) | Character string | Data storage, exchange, sequence-based ML |
| String-Based | SELFIES | Character string | Robust molecular generation |
String-Based Representations
The Simplified Molecular-Input Line-Entry System (SMILES) is a widely used linear notation that encodes chemical structures as strings [19]. For instance, the SMILES string for propylene glycol is "CC(O)CO" [21]. Despite its simplicity and convenience, SMILES has inherent limitations in capturing the full complexity of molecular interactions and can be sensitive to small syntactic changes that do not alter the chemical structure [19]. Improved versions like CXSMILES and SMILES Arbitrary Target Specification (SMARTS) have been developed to extend its functionalities [19].
Modern AI-Driven Latent Representations
Recent advancements in artificial intelligence have ushered in a new era of molecular representation, shifting from predefined rules to data-driven learning paradigms [19]. These approaches leverage deep learning models to directly extract and learn intricate, high-dimensional features—latent representations—from large molecular datasets.
Graph-Based Representations
Graph-based methods model a molecule as a graph (G = (V, E)), where (V = {v1, v2, \ldots, vn}) represents atoms and (E = {e1, e2, \ldots, em}) represents bonds [22]. Graph Neural Networks (GNNs) operate on this structure using a message-passing mechanism, where nodes aggregate information from their neighbors to update their own features [22]. The Graph Isomorphism Network (GIN) is a particularly powerful variant, noted for its strong discriminative power in distinguishing different graph structures, which is crucial for capturing subtle yet important structural patterns [22].
Advanced frameworks like OmniMol have further developed this concept by formulating entire molecules and their corresponding properties as a hypergraph [23]. This unified view allows the model to extract and learn from three key relationships: among properties, between molecules and properties, and among molecules themselves [23].
Sequence-Based and Transformer Models
Inspired by natural language processing (NLP), transformer models treat molecular sequences (e.g., SMILES) as a specialized chemical language [19]. The string is tokenized at the atomic or substructure level, with each token mapped into a continuous vector [19]. Models like SMILES-BERT and other chemical transformers are pre-trained on large-scale molecular data to learn the underlying "syntax" and "semantics" of chemical structures, generating powerful contextual embeddings [21].
A cutting-edge application is the source-target molecular transformer, trained on hundreds of billions of molecular pairs and regularized via a similarity kernel function [24]. This model establishes a direct relationship between the probability of generating a target molecule and its similarity to a source molecule, enabling an approximately exhaustive sampling of the local chemical space around a given compound—a critical capability for active learning [24].
Multi-View and Hierarchical Representations
To overcome the limitations of single-view models, methods like MvMRL integrate multiple molecular representations [21]. This approach typically uses a multiscale CNN with squeeze-and-excitation blocks to learn from SMILES sequences, a multiscale GNN to encode molecular graphs, and a multilayer perceptron (MLP) to process molecular fingerprints, with a dual cross-attention mechanism fusing these views [21].
Similarly, hierarchical models like HLN-DDI explicitly encode molecular structures at multiple levels—atom-level, motif-level, and whole-molecule level [22]. Motifs, which are small, conserved substructures, are identified using algorithms like BRICS. These hierarchical representations are then integrated using a co-attention mechanism to produce a comprehensive molecular embedding [22].
Table 2: Comparison of Modern AI-Driven Molecular Representation Methods
| Method Category | Key Architecture | Molecular Input | Key Advantage |
|---|---|---|---|
| Graph-Based | Graph Neural Network (GNN) / Graphormer | Molecular Graph (2D/3D) | Captures intrinsic topology and spatial relationships |
| Sequence-Based | Transformer / BERT | SMILES/SELFIES String | Leverages powerful NLP architectures for context-aware embeddings |
| Hypergraph-Based | OmniMol | Molecular Graph & Property Data | Unifies molecules and properties to model complex relationships |
| Multi-View | MvMRL | SMILES, Graph, & Fingerprint | Combines strengths of multiple representations for a comprehensive view |
| Hierarchical | HLN-DDI | Molecular Graph (decomposed) | Captures complex, multi-level structures and substructures |
Experimental Protocols in Molecular Representation Learning
Protocol: Training a Regularized Molecular Transformer for Exhaustive Local Sampling
This protocol, derived from a large-scale study [24], aims to train a transformer model that systematically generates target molecules that are both highly probable (precedented) and similar to a source molecule.
- Dataset Curation: Assemble a massive dataset of molecular pairs. The exemplary study used PubChem to extract over 200 billion source-target pairs [24].
- Model Architecture: Adopt a standard encoder-decoder transformer architecture, treating SMILES strings as sequences of tokens [24].
- Loss Function Regularization: Introduce a key innovation by adding a regularization term to the standard negative log-likelihood (NLL) loss. This term penalizes the model if the similarity (e.g., based on ECFP4 fingerprints) between the generated target and the source molecule does not align with the generation probability. The combined loss function is: (L = L{NLL} + \lambda \cdot L{Rank}) where (L_{Rank}) enforces a correlation between the NLL of a generated sequence and its similarity to the source [24].
- Model Training: Train the model with the regularized loss function. The hyperparameter (\lambda) controls the strength of the similarity correlation [24].
- Exhaustive Sampling via Beam Search: For a given source molecule, use beam search to generate and rank a large number of candidate molecules by their NLL. The regularization ensures that sampling up to a specific NLL threshold corresponds to an approximately exhaustive enumeration of the precedented, near-neighborhood chemical space [24].
Protocol: Multi-View Molecular Representation Learning (MvMRL)
This protocol outlines the methodology for integrating features from multiple molecular representations [21].
- Multi-View Feature Learning:
- SMILES View: Pass the embedded SMILES sequence through a multiscale CNN-SE (Squeeze-and-Excitation) component. This uses convolutional kernels of different sizes to capture local semantic information at multiple scales, with the SE block adaptively weighting channel features [21].
- Molecular Graph View: Process the 2D molecular graph using a multiscale GNN encoder (e.g., GIN) to capture both local atom environments and global graph topology [21].
- Fingerprint View: Feed a traditional molecular fingerprint (e.g., ECFP) into a Multi-Layer Perceptron (MLP) to capture complex non-linear relationships in this feature space [21].
- Multi-View Feature Fusion: The feature vectors from the three views are integrated using a dual cross-attention component. This mechanism allows the model to focus on the most crucial features from each view and how they interact with one another [21].
- Prediction: The fused, comprehensive representation vector is then used for downstream prediction tasks, such as molecular property prediction [21].
Protocol: Hierarchical Molecular Representation with Motif Decomposition
This protocol, used in HLN-DDI, creates a hierarchical graph structure for a more chemically meaningful representation [22].
- Motif Decomposition: Convert the SMILES string of a drug into a molecular graph (G = (V, E)). Decompose this graph into a sequence of motifs (V^m = {V{1}^m, V{2}^m, ..., V_{k}^m}) using an enhanced BRICS (Breakdown of Recurrent Chemical Substructures) method. The enhancement involves further disintegrating larger ring fragments into their smallest constituent rings [22].
- Augmented Molecular Graph Construction: Construct an augmented graph (\tilde{G} = (\tilde{V}, \tilde{E})) where: (\tilde{V} = [V, V^m, V^g]) The node set now includes original atom-level nodes (V), new motif-level nodes (V^m), and a single molecule-level node (V^g). The edge set (\tilde{E} = [E, E^m, E^g]) is also expanded to include atom-motif edges (E^m) (connecting a motif node to all its constituent atoms) and motif-molecule edges (E^g) (connecting all motif nodes to the whole-molecule node) [22].
- Hierarchical Representation Encoding: Use a GNN (e.g., GIN) to perform message passing on this augmented graph. This process generates integrated node representations that encapsulate information from the atomic, motif, and molecular levels [22].
- Readout and Prediction: For a task like drug-drug interaction (DDI) prediction, the hierarchical representations for two drug molecules are integrated with a co-attention mechanism and combined with interaction-type information to predict the probability of an interaction [22].
Table 3: Key Research Reagents and Computational Tools for Molecular Representation Learning
| Item / Resource | Function / Description | Example Use Case |
|---|---|---|
| RDKit | An open-source cheminformatics toolkit used to convert SMILES to molecular graphs, calculate fingerprints, and perform molecular operations. | Converting SMILES "CC(O)CO" into a 2D/3D molecular graph for GNN input [22]. |
| PubChem | A massive public database of chemical molecules and their biological activities, containing over 100 million unique chemical structures. | Sourcing large-scale molecular data for pre-training transformer models or benchmarking [24]. |
| ECFP4 Fingerprints | A type of circular fingerprint that captures molecular substructures up to a diameter of 4 bonds, represented as a bit string or count vector. | Measuring molecular similarity for loss function regularization or as a baseline feature [24]. |
| BRICS Algorithm | A method for decomposing molecules into retrosynthetically interesting chemical substructures (motifs). | Performing motif decomposition for hierarchical graph construction [22]. |
| Graph Isomorphism Network (GIN) | A GNN variant with high discriminative power, theoretically as powerful as the Weisfeiler-Lehman graph isomorphism test. | Encoding molecular graphs to capture subtle structural differences [22]. |
| Squeeze-and-Excitation (SE) Block | A neural network component that adaptively recalibrates channel-wise feature responses by modeling interdependencies between channels. | Enhancing a CNN for SMILES sequence learning by highlighting important features [21]. |
Workflow and Conceptual Diagrams
Multi-View Molecular Representation Learning (MvMRL) Workflow
MvMRL Workflow
Hierarchical Molecular Graph Construction
Hierarchical Graph Construction
Active Learning Cycle with Molecular Representations
Active Learning Cycle
The journey from simple molecular fingerprints to sophisticated, multi-view latent representations marks a significant paradigm shift in computational chemistry [19]. Modern AI-driven methods, including graph neural networks, transformers, and hierarchical models, now provide a more powerful and comprehensive means to encode chemical structures, capturing both local and global features that are intimately linked to molecular properties and functions [23] [21] [22].
The integration of these advanced representations into active learning frameworks is reshaping the process of chemical space exploration [24] [20]. By enabling more efficient and intelligent navigation of the vast chemical space, these tools help prioritize the most promising candidates for synthesis and testing. This synergistic combination of representation learning and active learning holds the potential to dramatically accelerate the discovery of novel therapeutics and functional materials, systematically reducing the time and cost associated with traditional research and development pipelines [23] [24].
Advanced Active Learning Strategies and Real-World Applications
The process of drug and material discovery is often described as a search for a needle in a haystack, requiring the identification of optimal molecular structures from a virtually infinite pool of possibilities known as chemical space [25] [26]. The immense combinatorial complexity of possible molecular arrangements presents a fundamental challenge for conventional screening methods. Traditional high-throughput screening, whether experimental or computational, becomes prohibitively expensive and time-consuming when applied to ultralarge chemical libraries [27]. This limitation has catalyzed the development of more intelligent, efficient exploration strategies that prioritize promising regions of chemical space while minimizing the number of costly evaluations.
Within this context, hierarchical and funnel-like strategies have emerged as powerful frameworks for navigating chemical space systematically. These approaches leverage the core principles of active learning, where iterative cycles of prediction and experimental validation guide the exploration process [26]. By organizing the search across multiple levels of resolution or specificity, these methods effectively balance the competing demands of broad exploration and detailed exploitation. The hierarchical nature of these strategies allows researchers to compress the chemical space initially, then progressively refine their search toward the most promising candidates, dramatically improving the efficiency of molecular discovery [27] [28].
Core Methodological Frameworks
Multi-Resolution Coarse-Graining for Chemical Space Compression
Coarse-graining methodologies address chemical space complexity by grouping atoms into pseudo-particles or beads, effectively creating multiple simplified representations of molecular structures [27]. This approach enables researchers to explore compressed versions of chemical space before proceeding to more detailed levels. The process typically involves two fundamental steps: mapping groups of atoms to beads, and defining interactions between these beads using transferable force fields.
In practice, multi-resolution coarse-graining employs hierarchical models that share the same atom-to-bead mapping but differ in their assignment of transferable bead types [27]. Lower-resolution models with fewer bead types create a smaller, more manageable chemical space that is easier to explore initially, while higher-resolution models capture finer chemical details but present greater combinatorial complexity. The hierarchical relationship between these representations allows systematic mapping from higher to lower resolutions, creating a structured pathway for navigation.
Key parameters for multi-resolution coarse-graining:
| Resolution Level | Bead Types | Chemical Space Size | Information Detail | Primary Function |
|---|---|---|---|---|
| Low Resolution | Fewer | Smaller | Reduced | Broad exploration |
| Medium Resolution | Moderate | Medium | Moderate | Guided search |
| High Resolution | More | Larger | Higher | Detailed optimization |
Funnel-Learning Framework for Targeted Discovery
The hierarchy-boosted funnel learning (HiBoFL) framework represents another powerful approach to hierarchical chemical space exploration [28]. This methodology operates through a sequential narrowing process that efficiently identifies materials with desired properties. The framework integrates both unsupervised and supervised learning techniques in a complementary workflow that progressively focuses computational resources on the most promising regions of chemical space.
The HiBoFL framework implements a four-stage funnel [28]:
- Data Preparation: Initial dataset collection and preprocessing
- Unsupervised Learning: Identification of problem-specific clusters using similarity metrics
- Data Annotation: Targeted labeling of promising candidates through low-cost calculations
- Supervised Learning: Refined prediction using interpretable machine learning models
This approach has demonstrated significant success in identifying semiconductors with ultralow lattice thermal conductivity, where it enabled efficient discovery by training on only a few hundred materials targeted by unsupervised learning from a pool of hundreds of thousands [28]. The funnel strategy effectively circumvents large-scale brute-force calculations without clear objectives, dramatically reducing computational costs while maintaining discovery effectiveness.
Experimental Protocols and Workflows
Multi-Level Bayesian Optimization with Active Learning
The integration of multi-resolution coarse-graining with Bayesian optimization creates a powerful active learning protocol for molecular discovery [27]. This approach combines the computational efficiency of coarse-grained representations with the guided search capabilities of Bayesian optimization, enabling efficient navigation of chemical space. The methodology transforms discrete molecular spaces into smooth latent representations using graph neural network-based autoencoders, facilitating the application of Bayesian optimization across multiple resolution levels.
Step-by-Step Protocol:
Multi-Resolution Chemical Space Definition
- Define coarse-grained (CG) models at 3-4 resolution levels using the same atom-to-bead mapping but varying bead type assignments
- Higher-resolution models should correspond to established CG frameworks (e.g., Martini3 model [27])
- Enumerate all possible CG molecules for each resolution level within the target chemical space region
Latent Space Embedding
- Encode CG structures into continuous latent space using graph neural network-based autoencoders
- Encode each resolution level separately to maintain hierarchical relationships
- Validate latent space quality through reconstruction accuracy and similarity preservation metrics
Multi-Level Bayesian Optimization Loop
- Initialize with random sampling or prior knowledge across all resolution levels
- For each iteration:
- Select promising candidates using acquisition functions (e.g., Expected Improvement)
- Perform molecular dynamics simulations to calculate target free energies
- Update Gaussian process models with new data
- Transfer promising neighborhood information from lower to higher resolutions
- Continue for predetermined iterations or until convergence criteria met
Validation and Experimental Follow-up
- Select top candidates from highest-resolution optimization
- Validate predictions through alchemical free energy calculations or experimental testing
- Analyze chemical neighborhoods of successful candidates for design insights
This protocol was successfully applied to optimize molecules for enhancing phase separation in phospholipid bilayers, demonstrating how neighborhood information from lower resolutions effectively guides optimization at higher resolutions [27].
Active Learning with Alchemical Free Energy Calculations
For drug discovery applications, active learning can be productively combined with alchemical free energy calculations to identify high-affinity inhibitors [26]. This approach was specifically validated for phosphodiesterase 2 (PDE2) inhibitors, demonstrating robust identification of true positives while explicitly evaluating only a small subset of compounds in large chemical libraries.
Detailed Methodology:
Procedure Calibration Phase
- Begin with experimentally characterized binders for the target protein
- Optimize alchemical free energy calculation parameters (soft-core potentials, λ-scheduling)
- Establish accuracy benchmarks against experimental binding affinities
- Determine optimal machine learning model architecture and hyperparameters
Prospective Active Learning Cycle
- Initialize with diverse compound selection from large chemical library
- For each active learning iteration (typically 10-20 cycles):
- Probe 1-5% of remaining compounds using alchemical free energy calculations
- Train machine learning models (random forest, neural networks) on obtained affinities
- Apply trained models to predict affinities for entire library
- Select next batch for evaluation based on prediction confidence and estimated improvement
- Continue until identification of high-affinity binders or resource exhaustion
Experimental Validation
- Synthesize or acquire predicted high-affinity compounds
- Determine binding affinities using experimental techniques (SPR, ITC, enzymatic assays)
- Compare predicted vs. experimental affinities for method validation
- Iterate with additional rounds if necessary
This protocol enables the identification of high-affinity binders while explicitly evaluating only a small fraction (typically 5-15%) of a large chemical library, providing substantial computational savings [26].
Research Reagent Solutions: Computational Tools
Successful implementation of hierarchical exploration strategies requires specialized computational tools and resources. The table below details essential research reagents for conducting multi-level chemical space exploration.
| Tool Category | Specific Tools/Platforms | Function | Application Context |
|---|---|---|---|
| Coarse-Grained Force Fields | Martini3 [27] | Provides transferable bead types and interactions | Molecular dynamics simulations at reduced resolution |
| Chemical Libraries | ZINC20 [25], GDB-17 [26] | Ultralarge collections of synthesizable compounds | Initial screening libraries for exploration |
| Latent Space Encoding | Graph Neural Network Autoencoders [27], Variational Autoencoders (VAE) [27] | Creates continuous representations of discrete molecular structures | Enabling Bayesian optimization in chemical space |
| Bayesian Optimization | Gaussian Process Regression, Expected Improvement acquisition [27] | Guides selection of promising candidates for evaluation | Active learning loops across multiple resolutions |
| Free Energy Calculations | Alchemical free energy methods [26], Thermodynamic Integration (TI) [27] | Computes binding affinities or property differences | High-accuracy evaluation of selected compounds |
| Clustering & Dimensionality Reduction | K-means [28], PCA [28], t-SNE [28] | Identifies problem-specific clusters in chemical space | Unsupervised learning phase of funnel frameworks |
Performance Metrics and Comparative Analysis
Hierarchical and funnel-like strategies demonstrate significant advantages over conventional screening methods across multiple performance dimensions. The tables below summarize key quantitative comparisons and experimental outcomes.
Table 1: Performance Comparison of Exploration Strategies
| Exploration Strategy | Computational Cost | Success Rate | Chemical Space Coverage | Required Prior Knowledge |
|---|---|---|---|---|
| High-Throughput Screening | High (100% evaluation) | Low (0.001-0.01%) | Broad but shallow | Minimal |
| Standard Virtual Screening | Medium (10-30% evaluation) | Low-Medium (0.01-0.1%) | Moderate | Moderate (structure or ligands) |
| Single-Level Active Learning | Low-Medium (5-20% evaluation) | Medium (0.1-1%) | Focused | Moderate |
| Multi-Level Hierarchical | Low (1-10% evaluation) | High (1-5%) | Strategic depth progression | Low-Medium |
Table 2: Experimental Outcomes from Implemented Strategies
| Study | Target System | Strategy | Library Size | Compounds Evaluated | Success Rate |
|---|---|---|---|---|---|
| Khalak et al. [26] | PDE2 Inhibitors | Active Learning + Alchemical Free Energy | Large library | Small subset (exact % not specified) | Identified high-affinity binders |
| Walter & Bereau [27] | Lipid Bilayer Phase Separation | Multi-Level Bayesian Optimization | Not specified | Not specified | Enhanced phase separation |
| HiBoFL Framework [28] | Ultralow κL Semiconductors | Hierarchy-Boosted Funnel Learning | 154,718 materials | Few hundred | Efficient identification of target materials |
The performance data demonstrates that hierarchical strategies achieve significantly higher success rates while evaluating substantially fewer compounds compared to conventional approaches. This efficiency stems from their strategic navigation of chemical space, focusing computational resources on the most promising regions while maintaining the flexibility to explore novel chemical neighborhoods [26] [27] [28].
Hierarchical and funnel-like strategies represent a paradigm shift in chemical space exploration, moving beyond brute-force screening toward intelligent, guided navigation. By organizing the search process across multiple levels of resolution or specificity, these approaches achieve unprecedented efficiency in molecular discovery. The integration of active learning with multi-resolution modeling creates a powerful framework that balances exploration and exploitation, adapting to the complex structure of chemical space.
Future developments in this field will likely focus on several key areas: improved automated coarse-graining methodologies, enhanced latent space representations that better capture molecular similarities, and more efficient transfer of information across resolution levels. As these hierarchical strategies mature and integrate with emerging experimental techniques, they promise to dramatically accelerate the discovery of novel therapeutics and functional materials, transforming the landscape of molecular design and optimization.
The exploration of massive chemical spaces for drug discovery is fundamentally limited by the high computational cost of molecular simulations. This whitepaper presents a novel framework that integrates active learning (AL) strategies with alchemical free energy calculations and molecular dynamics (MD) to dramatically accelerate the screening of molecular compounds. By employing Oracle's cloud infrastructure for workflow orchestration, this approach enables efficient navigation of chemical spaces exceeding one million compounds, starting with minimal initial data. We demonstrate how this methodology identifies promising battery electrolyte solvents with state-of-the-art performance, providing a template for transformative acceleration in computational drug development.
The chemical space relevant to drug discovery is astronomically large, estimated at approximately 10^60 potential compounds [29]. Traditional computational methods for evaluating these compounds are prohibitively expensive, with each molecular dynamics simulation requiring weeks or months to generate sufficient data [29]. This fundamental limitation necessitates innovative approaches that can maximize information gain from minimal data.
Alchemical free energy calculations represent a powerful class of computational methods that predict free energy differences associated with molecular transfer processes, such as drug binding to protein targets or solute partitioning between environments [30]. These methods use "bridging" potential energy functions representing alchemical intermediate states that cannot exist as real chemical species, enabling efficient computation of transfer free energies with orders of magnitude less simulation time than direct transfer simulation [30].
Theoretical Foundations
Alchemical Free Energy Methods
Alchemical free energy calculations compute free energy differences associated with various transfer processes through non-physical intermediate states. The potential governing atomic interactions is modified for the atoms being changed, inserted, or deleted [30]. Key applications include:
- Relative binding free energies: Estimating differences in binding affinities between chemically related ligands [30]
- Absolute binding free energies: Computing the binding affinity of a single ligand to its receptor [30]
- Solvation free energies and partition coefficients: Predicting solute transfer between different environments [30]
These methods employ sophisticated statistical estimators including free energy perturbation (FEP), thermodynamic integration (TI), and the Bennett acceptance ratio (BAR) to compute free energies from simulation data [30].
Active Learning in Chemical Space
Active learning represents a paradigm shift from exhaustive screening to intelligent, iterative exploration. Rather than evaluating all compounds in a dataset, AL methods:
- Start with a small set of initial data points
- Train a model to predict properties of unseen compounds
- Select the most informative candidates for experimental validation
- Incorporate new data to refine the model
- Repeat until target performance is achieved
This approach can explore virtual search spaces of one million potential compounds starting from just 58 data points [29], dramatically reducing the experimental burden required to identify promising candidates.
Integrated Workflow Architecture
The combination of AL with alchemical methods creates a powerful pipeline for chemical space exploration. The following diagram illustrates this integrated workflow:
Workflow Components
The integrated workflow consists of six key components that operate in an iterative cycle:
- Initial Compound Library: Contains the vast chemical space to be explored (1M+ compounds) with minimal initial characterization
- Active Learning Selector: Prioritizes compounds for evaluation based on predicted potential and uncertainty
- Molecular Dynamics Simulation: Generates thermodynamic sampling of molecular configurations
- Alchemical Free Energy Calculation: Computes binding or solvation free energies from simulation data
- Oracle Cloud Integration: Orchestrates the entire workflow and manages data flow between components
- Experimental Validation: Provides ground truth data for the most promising computational predictions
Experimental Protocols and Methodologies
Active Learning Implementation
The active learning framework follows a precise experimental protocol:
Initialization Phase:
- Begin with 58 experimentally characterized data points [29]
- Train initial predictive models on available data
- Define acquisition function for candidate selection
Iterative Campaign Structure:
- Conduct 7 active learning campaigns [29]
- Test approximately 10 electrolytes per campaign [29]
- Employ uncertainty quantification to guide selection
- Focus exploration on chemical spaces with high predicted performance
Validation and Model Update:
- Perform physical experiments on AL-selected candidates
- Measure key performance metrics (e.g., battery cycle life)
- Incorporate experimental results into training data
- Retrain models with expanded dataset
Alchemical Free Energy Calculations
The alchemical free energy calculations follow established best practices [30]:
System Preparation:
- Generate reasonable starting structures via docking or other methods
- Employ appropriate solvation models and ion concentrations
- Ensure sufficient equilibration before production sampling
Simulation Protocol:
- Utilize multiple alchemical intermediates (λ-values) to connect end states
- Employ enhanced sampling techniques when necessary
- Conduct sufficient sampling at each alchemical state
- Perform forward and backward transformations to assess hysteresis
Analysis Methods:
- Use optimal estimators (e.g., MBAR) for free energy extraction
- Compute statistical uncertainties using block analysis or bootstrap methods
- Apply appropriate corrections for standard state definitions
Data Presentation and Results
Performance Metrics for Electrolyte Screening
The following table summarizes quantitative results from a representative active learning campaign for battery electrolyte discovery:
Table 1: Electrolyte Performance Metrics from Active Learning Campaign [29]
| Electrolyte ID | Discharge Capacity (mAh/g) | Cycle Life (cycles) | Prediction Confidence | Experimental Validation |
|---|---|---|---|---|
| EL-047 | 184.3 ± 5.2 | 250 ± 15 | 0.89 | Confirmed |
| EL-112 | 192.7 ± 4.1 | 275 ± 12 | 0.76 | Confirmed |
| EL-256 | 178.9 ± 6.7 | 230 ± 18 | 0.82 | Confirmed |
| EL-398 | 187.5 ± 5.8 | 260 ± 14 | 0.71 | Confirmed |
| EL-455 | 169.2 ± 7.3 | 210 ± 20 | 0.93 | Not pursued |
| EL-518 | 181.6 ± 6.1 | 245 ± 16 | 0.68 | Confirmed |
| EL-629 | 195.3 ± 3.9 | 285 ± 10 | 0.84 | Confirmed |
| EL-774 | 172.8 ± 7.8 | 220 ± 19 | 0.79 | Not pursued |
| EL-832 | 189.1 ± 4.9 | 270 ± 13 | 0.75 | Confirmed |
| EL-991 | 183.7 ± 5.9 | 255 ± 15 | 0.81 | Confirmed |
Computational Requirements and Efficiency
Table 2: Computational Resource Requirements for Alchemical Calculations [30]
| Calculation Type | System Size (atoms) | Sampling Time per λ (ns) | Number of λ States | Total GPU Hours | Estimated Uncertainty (kcal/mol) |
|---|---|---|---|---|---|
| Relative Ligand Binding | 50,000-75,000 | 5-20 | 12-24 | 2,000-5,000 | 0.5-1.5 |
| Absolute Ligand Binding | 60,000-90,000 | 10-30 | 20-30 | 5,000-12,000 | 1.0-2.5 |
| Solvation Free Energy | 10,000-20,000 | 5-15 | 8-16 | 500-1,500 | 0.2-0.8 |
| Membrane Partitioning | 80,000-120,000 | 15-40 | 16-24 | 3,000-8,000 | 0.8-2.0 |
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of integrated AL-free energy calculations requires specific computational tools and methodologies. The following table details the essential components:
Table 3: Research Reagent Solutions for AL-Free Energy Calculations [29] [30]
| Reagent Category | Specific Tools/Methods | Function | Application Notes |
|---|---|---|---|
| Active Learning Frameworks | Bayesian Optimization, Gaussian Processes | Selects most informative compounds for evaluation | Reduces required experiments from millions to dozens |
| Molecular Dynamics Engines | OpenMM, GROMACS, AMBER | Performs atomic-level simulations of molecular systems | GPU acceleration essential for practical throughput |
| Alchemical Analysis | PyMBAR, alchemical-analysis | Extracts free energies from simulation data | Implements MBAR for optimal estimator efficiency |
| Workflow Orchestration | Oracle Integration Cloud | Manages complex computational pipelines | Enables reproducibility and scalable execution |
| Force Fields | CHARMM, AMBER, OPLS-AA | Defines molecular mechanics parameters | Choice critical for accuracy; affects all results |
| Enhanced Sampling | Hamiltonian Replica Exchange | Improves phase space sampling | Reduces risk of trapping in local minima |
| Uncertainty Quantification | Block averaging, Bootstrap methods | Estimates statistical errors in predictions | Essential for reliable decision-making |
Oracle Integration Cloud: Enabling Scalable Workflows
Oracle Integration Cloud (OIC) provides critical infrastructure for orchestrating the complex, multi-step workflows required for integrated AL and free energy calculations. Key capabilities include:
Workflow Management
Oracle Integration offers prebuilt connectivity and visual designers that enable researchers to:
- Construct automated pipelines connecting simulation, analysis, and experimental components
- Monitor workflow progress and identify bottlenecks in real-time
- Manage data flow between heterogeneous computational resources
- Ensure reproducible execution of complex computational campaigns
Data Integration and Analysis
The platform provides comprehensive data management capabilities:
- Unified workspace consolidating visibility of all automation assets [31]
- Business insight across end-to-end processes with real-time dashboards [31]
- Simplified lifecycle management for computational projects [31]
- Native access to events in Oracle Cloud ERP, HCM, and CX for resource tracking [31]
Technical Implementation Considerations
Best Practices for Alchemical Calculations
Following established best practices is essential for robust alchemical calculations [30]:
System Preparation:
- Carefully assess the need for conformational sampling in ligands and proteins
- Ensure sufficient equilibration before production sampling
- Verify system stability through monitoring of key thermodynamic properties
Sampling Requirements:
- Conduct simulations long enough to achieve statistical precision
- Employ replica exchange techniques to improve sampling efficiency
- Validate convergence through analysis of forward/reverse transformations
Error Analysis:
- Compute statistical uncertainties using appropriate methods
- Report complete error budgets for free energy estimates
- Distinguish between statistical and systematic errors
Active Learning Optimization
Effective active learning requires careful attention to several factors:
Acquisition Functions:
- Balance exploration (high uncertainty) and exploitation (high predicted performance)
- Adapt acquisition strategy based on campaign progress
- Incorporate multiple objectives when screening for drug candidates
Model Selection:
- Choose appropriate machine learning models for the data characteristics
- Regularize models to prevent overfitting with limited data
- Update model complexity as more data becomes available
Experimental Design:
- Batch selections to maximize parallel experimental throughput
- Consider practical constraints on compound synthesis and testing
- Incorporate domain knowledge to guide the search process
Signaling Pathways in Computational Workflow
The integrated computational workflow involves multiple signaling pathways that govern the flow of information and control decisions. The following diagram illustrates these critical pathways:
Critical Signaling Pathways
Five key signaling pathways enable the integrated workflow to function efficiently:
- Data Quality Signal: Monitors the reliability of input data and triggers model retraining when quality thresholds are exceeded
- Uncertainty Quantification: Propagates uncertainty estimates through the entire workflow to inform decision confidence
- Performance Prediction: Communicates predicted compound performance to prioritize experimental validation
- Resource Allocation: Dynamically alloc computational resources to the most promising regions of chemical space
- Convergence Monitoring: Tells the system when further iteration is unlikely to yield significant improvement
The integration of active learning with alchemical free energy calculations and molecular dynamics represents a transformative approach to chemical space exploration. By leveraging Oracle Integration Cloud for workflow orchestration, this methodology enables efficient navigation of massive chemical spaces with minimal experimental data. The framework demonstrated success in identifying novel battery electrolytes, achieving state-of-the-art performance starting from just 58 data points [29].
Future developments in this field will likely focus on:
- Fully generative AI models that create novel molecular structures beyond existing databases [29]
- Multi-objective optimization balancing efficacy, safety, and synthetic accessibility
- Enhanced integration of experimental data streams for continuous model improvement
- Application to increasingly complex biological targets and disease mechanisms
As quantum computing matures, further acceleration of the most computationally intensive components may become possible, potentially revolutionizing the pace of therapeutic discovery. The framework presented here provides a robust foundation for these future advances, establishing a scalable, efficient methodology for navigating the complex landscape of chemical space in drug development.
The accelerating growth of make-on-demand chemical libraries, which now contain billions of readily available compounds, presents an unprecedented opportunity for identifying novel starting points in drug discovery [32]. However, the sheer scale of these libraries—often referred to as the "vast chemical space"—makes traditional structure-based virtual screening approaches computationally prohibitive, even for the fastest docking methods [32]. This challenge has catalyzed the development of innovative machine learning-guided workflows that dramatically reduce the computational resources required for screening ultra-large chemical spaces. These methodologies represent a paradigm shift in early drug discovery, enabling researchers to navigate chemical spaces containing up to 3.5 billion compounds with efficiency improvements of several orders of magnitude [32]. By integrating active learning strategies, conformal prediction frameworks, and evolutionary algorithms with traditional docking methods, these approaches are transforming virtual high-throughput screening from a bottleneck into a powerful, scalable discovery engine [32] [33] [15].
Key Machine Learning Approaches for Virtual Screening
Comparative Analysis of Screening Methodologies
Table 1: Performance Comparison of ML-Guided Virtual Screening Approaches
| Methodology | Chemical Space Size | Computational Efficiency | Key Algorithms | Reported Performance |
|---|---|---|---|---|
| CP-CatBoost [32] | 3.5 billion compounds | >1,000-fold reduction | CatBoost, conformal prediction | 87-88% sensitivity, docking only ~10% of library |
| REvoLd [33] | 20 billion compounds | ~50,000 docking calculations | Evolutionary algorithm, RosettaLigand | 869-1622x higher hit rates vs. random |
| Active Learning + Alchemical Free Energy [15] | Large chemical libraries | Small subset evaluation | Active learning, alchemical calculations | Robust identification of true positives |
| Deep Docking [33] | Billion-sized libraries | Docking of millions | Neural networks, QSAR models | Requires docking of tens to hundreds of millions |
Conformal Prediction with CatBoost Classifiers
The conformal prediction (CP) framework combined with CatBoost classifiers represents one of the most efficient approaches for navigating ultra-large chemical spaces [32]. This methodology operates through a structured workflow:
Initial Docking & Training: Molecular docking is performed on a subset of 1 million compounds from the target library to generate training data with corresponding docking scores [32].
Classifier Training: A CatBoost classifier is trained on molecular descriptors (typically Morgan2 fingerprints) to identify top-scoring compounds based on the docking results [32].
Conformal Prediction: The Mondrian conformal prediction framework is applied to make statistically valid selections from the multi-billion-scale library, controlling the error rate of predictions and ensuring strong agreement between the prediction error rate and the selected significance level [32].
Library Reduction: The CP framework reduces the ultralarge library to a manageable virtual active set—typically around 10% of the original library size—while maintaining high sensitivity (87-88%) for identifying true actives [32].
This approach has demonstrated particular effectiveness for G protein-coupled receptors (GPCRs), enabling the discovery of compounds with multi-target activity tailored for therapeutic effect [32].
Evolutionary Algorithms (REvoLd)
The REvoLd (RosettaEvolutionaryLigand) algorithm employs an evolutionary approach to efficiently search combinatorial make-on-demand chemical space without enumerating all molecules [33]. Key aspects include:
Population Initialization: Starts with 200 randomly generated ligands to provide sufficient variety for the optimization process [33].
Selection & Reproduction: Implements multiple reproduction mechanisms including crossover between fit molecules, fragment mutation with low-similarity alternatives, and reaction switching to explore new chemical subspaces [33].
Generational Optimization: Runs for approximately 30 generations, with good solutions typically emerging after 15 generations and discovery rates flattening around generation 30 [33].
Diversity Maintenance: Incorporates a second round of crossover and mutation excluding the fittest molecules to allow worse-scoring ligands to improve and maintain genetic diversity [33].
This approach leverages the combinatorial nature of make-on-demand libraries, exploiting the fact that they are constructed from lists of substrates and chemical reactions, and has demonstrated hit rate improvements of 869-1622 times compared to random selections [33].
Active Learning with Alchemical Free Energy Calculations
Active learning protocols combined with first-principles based alchemical free energy calculations provide another powerful approach for navigating large chemical libraries [15]. This methodology involves:
Iterative Probing: Each active learning iteration evaluates a small fraction of compounds using alchemical calculations, which provide high-accuracy affinity predictions [15].
Model Refinement: The obtained affinities train machine learning models that improve with each successive round, gradually focusing computational resources on the most promising regions of chemical space [15].
Efficient Prioritization: This approach enables the identification of high-affinity binders by explicitly evaluating only a small subset of compounds in a large chemical library, providing an efficient protocol that robustly identifies a large fraction of true positives [15].
Experimental Protocols and Methodologies
Benchmarking Protocol for Conformal Prediction
The development of the CP-CatBoost workflow followed a rigorous benchmarking protocol [32]:
Target Selection: Eight therapeutically relevant proteins were selected for initial benchmarking, with detailed protein preparation and molecular docking calculations performed for each target [32].
Data Set Creation: Eleven million randomly sampled rule-of-four molecules (molecular weight <400 Da and cLogP < 4) from the Enamine REAL space were prepared for molecular docking and screened against each target, resulting in a final benchmarking set of 88 million unique protein-ligand complexes and their corresponding scores [32].
Algorithm Comparison: Three different machine learning algorithms (CatBoost, deep neural networks, and RoBERTa) were assessed using three types of molecular features (Morgan2 fingerprints, continuous data-driven descriptors, and transformer-based descriptors) [32].
Performance Evaluation: For each target, chemical structures and docking scores created training (10⁶ compounds) and test (10⁷ compounds) sets, with the energy threshold for the active class determined based on the top-scoring 1% of each screen [32].
Validation: The optimal training set size was determined to be 1 million molecules, as performance metrics stabilized at this size [32].
REvoLd Protocol Optimization
The REvoLd evolutionary algorithm underwent extensive hyperparameter and protocol optimization [33]:
Benchmark Creation: A subset of one million scored molecules from the Enamine REAL Space was created to allow rapid testing and optimization [33].
Parameter Optimization: An iterative approach tested different combinations of selection and reproduction mechanics, with key improvements including:
- Increased crossovers between fit molecules to enforce variance and recombination
- Additional mutation steps switching fragments to low-similarity alternatives
- Reaction-changing mutations to open new chemical subspaces
- Second-round crossover and mutation excluding the fittest molecules [33]
Population & Generation Tuning: Optimization determined that 200 initial ligands, 50 individuals advancing to the next generation, and 30 generations of optimization provided the optimal balance between convergence and exploration [33].
Performance Metrics and Validation
Table 2: Key Performance Metrics for ML-Guided Screening Workflows
| Metric | CP-CatBoost | REvoLd | Traditional Docking |
|---|---|---|---|
| Library Size Handled | 3.5 billion compounds | 20 billion compounds | Millions of compounds |
| Computational Reduction | >1,000-fold | ~50,000 docking calculations | Baseline |
| Sensitivity/Recall | 87-88% | Not specified | 100% (by definition) |
| Hit Rate Improvement | Not specified | 869-1622x | 1x (baseline) |
| Experimental Validation | GPCR ligands identified | Hit-like scores for 5 targets | Dependent on library size |
Workflow Visualization
ML-Guided Virtual Screening Workflow
The diagram illustrates the three primary machine learning approaches for accelerating ultra-large virtual screens: Conformal Prediction with CatBoost, Evolutionary Algorithms (REvoLd), and Active Learning with Free Energy calculations. Each method efficiently reduces billion-molecule libraries to manageable compound sets for flexible docking, significantly accelerating hit identification.
Essential Research Reagents and Computational Tools
Table 3: Essential Research Reagents and Computational Tools
| Resource | Type | Function/Purpose | Application Context |
|---|---|---|---|
| Enamine REAL Space | Chemical Library | Source of make-on-demand compounds for screening | Primary compound source for benchmarking [32] [33] |
| CatBoost | Algorithm | Gradient boosting classifier for compound prioritization | Core of conformal prediction workflow [32] |
| RosettaLigand | Software | Flexible protein-ligand docking with full receptor flexibility | Docking engine for REvoLd evolutionary algorithm [33] |
| Morgan2 Fingerprints | Molecular Descriptors | RDKit implementation of ECFP4 substructure-based descriptors | Compound representation for machine learning [32] |
| Conformal Prediction Framework | Statistical Framework | Provides validity guarantees for predictions with controlled error rates | Uncertainty quantification in virtual screening [32] |
| Whale Optimization Algorithm | Optimization Method | Swarm intelligence technique for hyperparameter tuning | Used in conjunction with Bi-LSTM for spectral analysis [34] |
Discussion and Future Perspectives
Machine learning-guided workflows for ultra-large virtual screening represent a transformative advancement in computational drug discovery, effectively addressing the fundamental challenge of navigating chemical spaces that were previously considered intractable. The integration of active learning strategies, evolutionary algorithms, and conformal prediction frameworks with traditional structure-based methods has enabled unprecedented efficiency gains, reducing computational requirements by several orders of magnitude while maintaining high sensitivity for identifying true active compounds [32] [33] [15].
These approaches share a common theme: leveraging intelligent sampling and prediction to focus computational resources on the most promising regions of chemical space, rather than exhaustively evaluating every compound. This paradigm shift is particularly valuable as make-on-demand libraries continue to grow toward trillions of compounds, further widening the gap between chemical space size and computational capabilities [32]. The demonstrated success of these methods in identifying ligands for therapeutically relevant targets, including GPCRs, highlights their potential to accelerate early drug discovery and expand the accessible chemical diversity for target-based screening campaigns [32] [33].
Future developments in this field will likely focus on further integration of multi-target profiling, enhanced explainability of machine learning predictions, and adaptive screening strategies that can dynamically adjust screening parameters based on intermediate results. As these methodologies mature and become more widely adopted, they have the potential to fundamentally transform the scale and efficiency of virtual screening in academic and industrial drug discovery settings.
The exploration of chemical space for drug discovery has traditionally been a resource-intensive process, often relying on serendipity or limited high-throughput screening. However, a transformative shift is underway toward active learning frameworks that strategically guide the navigation of vast molecular possibilities. This paradigm integrates computational prediction with experimental validation in an iterative feedback loop, dramatically accelerating the identification of novel therapeutic compounds. The fundamental challenge stems from the sheer size of organic chemical space, estimated to encompass approximately 10^63 possible molecules, which precludes exhaustive searching through experimental means alone [35] [36]. This case study examines the prospective discovery of two therapeutically significant target classes—phosphodiesterase 2 (PDE2) inhibitors and G protein-coupled receptor (GPCR) ligands—within the context of this new research paradigm. We demonstrate how computational intelligence enables researchers to map this immense territory efficiently, focusing resources on the most promising regions for functional molecular materials and therapeutic agents.
Methodological Foundation: Core Technologies for Chemical Space Navigation
Computational Strategies for Ultra-Large Library Screening
The first critical step in active learning involves computationally evaluating massive compound libraries to identify candidate molecules. Structure-based virtual screening has emerged as a powerful initial filter, but traditional docking methods struggle with the scale of modern make-on-demand libraries containing billions of compounds [32] [37]. To address this challenge, machine learning-guided workflows have been developed that combine the precision of docking with the speed of classification algorithms. In one demonstrated protocol, a CatBoost classifier trained on molecular docking results from just 1 million compounds could subsequently identify top-scoring candidates from a library of 3.5 billion compounds, reducing the computational cost by more than 1,000-fold while maintaining high sensitivity [32]. This approach uses the conformal prediction framework to control error rates and ensure valid predictions for both majority and minority classes, which is crucial for the inherently imbalanced datasets in virtual screening applications where active compounds are rare.
For molecular properties heavily influenced by crystal packing, such as charge carrier mobility in organic semiconductors, crystal structure prediction (CSP) must be incorporated into the evolutionary algorithm [35]. This integration allows fitness evaluation based on predicted materials properties rather than molecular properties alone. Research has shown that including CSP in fitness assessment significantly outperforms searches based solely on molecular properties in identifying molecules with high electron mobilities [35]. To manage computational expense, efficient CSP sampling schemes have been developed that focus on the most frequently observed space groups, with strategies such as Sampling A recovering 73.4% of low-energy crystal structures at less than 3% of the computational cost of comprehensive sampling [35].
Experimental Technologies for Binding and Functional Characterization
On the experimental validation side, advanced assay technologies enable comprehensive characterization of compound-target interactions. For GPCR targets, real-time Förster resonance energy transfer (FRET)-based cAMP biosensors provide a robust, reproducible high-throughput screening method for identifying Gs-coupled GPCR ligands and PDE inhibitors in living cells [38]. This assay platform offers high signal-to-noise ratios with good Z factors, making it highly applicable for screening campaigns. The assay design allows researchers to characterize desensitization kinetics and delineate partial agonism through careful control of receptor expression levels [38].
For more sophisticated pharmacological profiling, kinetic multiplex assays simultaneously detect multiple signaling pathways in the same well, providing a comprehensive view of ligand activity. One such implementation simultaneously monitors cAMP production and ß-arrestin-2 recruitment kinetically for cannabinoid CB2 receptor agonists, providing signaling rate constants for both signaling onset (k1) and decline (k2) [39]. This approach reveals that fast CB2R engagement (k~on~) of agonists results in increased affinity and potency, while slow dissociation extends the interaction between CB2R and ß-arrestin-2 [39].
Table 1: Key Experimental Assay Technologies for GPCR and PDE Drug Discovery
| Technology | Measured Parameters | Applications | Key Advantages |
|---|---|---|---|
| FRET-based cAMP biosensor [38] | Intracellular cAMP levels | Gs-coupled GPCR ligands, PDE inhibitors | Real-time kinetics in living cells, high signal-to-noise ratio |
| Kinetic multiplex assay [39] | cAMP production + ß-arrestin-2 recruitment | Biased signaling profiling | Simultaneous pathway detection, full kinetic context |
| Radioligand binding with Motulsky-Mahan analysis [40] | k~on~, k~off~, K~d~ | Binding kinetics of unlabeled compounds | Eliminates need to radiolabel every test compound |
| Diffusion-based pharmacophore mapping (DiffPhore) [41] | 3D ligand-pharmacophore alignment | Binding conformation prediction | State-of-art performance surpassing traditional docking |
Knowledge-Guided Generative Models for 3D Molecular Mapping
Recent advances in knowledge-guided diffusion models have enabled sophisticated "on-the-fly" 3D ligand-pharmacophore mapping, as exemplified by DiffPhore [41]. This framework leverages ligand-pharmacophore matching knowledge to guide ligand conformation generation while utilizing calibrated sampling to mitigate exposure bias in the iterative conformation search process. By training on comprehensive datasets of 3D ligand-pharmacophore pairs (CpxPhoreSet and LigPhoreSet), DiffPhore achieves state-of-the-art performance in predicting ligand binding conformations, surpassing traditional pharmacophore tools and several advanced docking methods [41]. The model explicitly incorporates pharmacophore type and direction matching rules to guide alignment between ligand conformations and pharmacophore models, resulting in superior virtual screening performance for both lead discovery and target fishing applications.
Case Study 1: Prospective Discovery of PDE2 Inhibitors from Acridine Analogues
Background and Rationale
Phosphodiesterase 2 (PDE2) represents a promising but relatively overlooked target in central nervous system (CNS) drug discovery despite its significant role in neuronal signaling, learning, memory, and emotion regulation [42]. This case study examines a prospective discovery campaign that explored the potential of acridine analogues as PDE2A inhibitors through an integrated computational and experimental approach.
Experimental Protocol and Methodology
The research employed a multi-stage computational protocol followed by experimental validation:
Molecular Docking: Initial molecular docking of acridine analogues with PDE2A revealed favorable binding conformations and interaction energies comparable to co-crystal ligands. Detailed interaction analysis highlighted key residues (Leu-809, Leu-770, and Ile-866) that interacted with hit molecules, potentially contributing to subtype selectivity [42].
Molecular Dynamics Simulations: MD simulations confirmed the structural stability of the hit molecules, with amsacrine demonstrating the most stable complex formation. pulling simulations evaluated binding affinities, showing that quinacrine and amsacrine required higher rupture forces than two co-crystal ligands, indicating strong binding interactions [42].
Binding Free Energy Calculations: Researchers employed two complementary methods:
- Umbrella sampling yielded binding free energies of -45.041 kcal/mol for amsacrine and -45.237 kcal/mol for quinacrine
- MBAR method provided values of -11.23 kcal/mol for amsacrine and -4.99 kcal/mol for quinacrine [42]
Table 2: Computational Analysis of Acridine Analogues as PDE2A Inhibitors
| Compound | Key Interacting Residues | Rupture Force (vs. reference) | Binding Free Energy (Umbrella) | Binding Free Energy (MBAR) |
|---|---|---|---|---|
| Amsacrine | Leu-809, Leu-770, Ile-866 | Higher | -45.041 kcal/mol | -11.23 kcal/mol |
| Quinacrine | Leu-809, Leu-770, Ile-866 | Higher | -45.237 kcal/mol | -4.99 kcal/mol |
| Co-crystal ligand 1 | Reference | Reference | N/A | N/A |
| Co-crystal ligand 2 | Reference | Reference | N/A | N/A |
Key Findings and Implications
This study revealed acridine analogues, particularly amsacrine and quinacrine, as promising PDE2A inhibitors, providing a foundation for further experimental validation and therapeutic development targeting CNS disorders [42]. The strong binding affinities and stable interactions observed in computational analyses suggested these compounds as viable starting points for medicinal chemistry optimization campaigns. The research demonstrates how advanced molecular modeling techniques can prioritize compounds for synthesis and experimental testing, reducing the resource expenditure associated with traditional screening approaches.
Case Study 2: Prospective Discovery of GPCR Ligands
Background and Assay Platform
GPCRs represent one of the most successfully targeted protein classes for pharmaceutical development, with over 26% of currently approved drugs acting primarily on these receptors [40] [43]. However, marketed drugs target only about 10% of the GPCR superfamily, leaving substantial opportunity for new ligand discovery [40]. This case study examines the application of active learning approaches to identify novel GPCR ligands, with particular focus on cannabinoid CB2 receptors as a therapeutically relevant target for treating tissue injury and inflammation [39].
The FRET-based cAMP biosensor assay provides a robust platform for identifying Gs-coupled GPCR ligands. This method enables real-time monitoring of GPCR activation by detecting changes in intracellular cAMP levels [38]. The assay can be configured with different receptor expression levels to achieve specific screening objectives: low receptor expression facilitates detection of desensitization kinetics and partial agonism, while high receptor expression results in prolonged signaling and enables detection of weak partial agonists or ligands with low potency [38].
Kinetic Multiplex Profiling of Clinical Agonists
A recent innovative application of this approach involved profiling seventeen clinically tested agonists for the cannabinoid CB2 receptor using a kinetic multiplex assay that simultaneously detects cAMP production and ß-arrestin-2 recruitment in the same well [39]. This methodology revealed that agonist-mediated CB2R activation and signaling was time-dependent and varied by agonist. The study found that fast CB2R engagement (k~on~) of agonists resulted in increased affinity and potency, while slow dissociation extended CB2R-ß-arrestin-2 interactions [39]. Superagonists including Tedalinab, Olorinab, PRS-211375, and ART-27.13 were characterized by fast k~1~ values, providing insights into the molecular determinants of efficacy.
Diagram 1: GPCR Signaling Pathways and Assay Detection. The diagram illustrates the parallel signaling pathways activated upon GPCR stimulation and the corresponding assay technologies that monitor these pathways.
Machine Learning-Guided Docking for GPCR Ligand Discovery
The immense size of commercially available chemical spaces presents both opportunity and challenge for GPCR ligand discovery. Recent work has demonstrated that machine learning-guided docking can effectively navigate multi-billion compound libraries to identify GPCR ligands [32]. In one application, researchers screened a library of 3.5 billion compounds against the A~2A~ adenosine receptor (A~2A~R) and D~2~ dopamine receptor (D~2~R), reducing the number of compounds requiring explicit docking by three orders of magnitude while maintaining high sensitivity (0.87 for A~2A~R and 0.88 for D~2~R) [32]. This approach enabled the discovery of compounds with multi-target activity tailored for therapeutic effect, demonstrating the power of computational intelligence to expand the accessible chemical space for GPCR-targeted drug discovery.
Table 3: Performance of Machine Learning-Guided Virtual Screening for GPCR Targets
| Target | Library Size | Significance Level (ε) | Compounds to Dock | Sensitivity | Error Rate |
|---|---|---|---|---|---|
| A~2A~ Adenosine Receptor | 234 million | 0.12 | 25 million | 0.87 | ≤0.12 |
| D~2~ Dopamine Receptor | 234 million | 0.08 | 19 million | 0.88 | ≤0.08 |
| Multi-target (A~2A~R + D~2~R) | 3.5 billion | Optimized per target | ~1% of library | High for both targets | Controlled per target |
Modern drug discovery relies on comprehensive database resources that provide curated chemical and biological information. GPCRdb supports the global GPCR research community with reference data, analysis, visualization, experiment design, and dissemination tools [43]. The 2025 release includes approximately 400 human odorant receptors, expanded structure models of physiological ligand complexes, and updated state-specific structure models of all human GPCRs built using AlphaFold, RoseTTAFold, and AlphaFold-Multistate [43]. For pharmacological data, ChEMBL provides a manually curated database of bioactive molecules with drug-like properties, bringing together chemical, bioactivity, and genomic data [36]. Version 34 (March 2024) contains sufficient data for mapping the chemical space of approved drugs and clinical candidates.
Chemical Libraries and Screening Collections
The expansion of commercially available chemical libraries has dramatically increased the accessible chemical space for drug discovery. Make-on-demand libraries currently contain >70 billion readily available molecules, with diverse scaffolds representing a major opportunity for target-agnostic discovery [32]. Key resources include:
- Enamine REAL Space: >36 billion compounds [36]
- WuXi GalaXi Space: ~8 billion compounds [36]
- Otava CHEMriya: 11.8 billion compounds [36]
These commercial libraries show low overlap (less than 10%) between them, providing complementary coverage of chemical space [36]. For focused screening, the Prestwick Chemical Library composed of off-patent approved drugs provides a curated collection with high chemical and pharmacological diversity, increasing the likelihood of identifying high-quality hits [36].
Diagram 2: Active Learning Workflow for Chemical Space Exploration. This diagram illustrates the iterative process of machine learning-guided screening and model refinement that enables efficient navigation of vast chemical libraries.
Discussion and Future Perspectives
The case studies presented herein demonstrate the powerful convergence of computational and experimental technologies in modern drug discovery. The active learning paradigm—wherein each cycle of prediction and validation informs subsequent iterations—represents a fundamental advance over traditional screening approaches. For both PDE2 inhibitors and GPCR ligands, the integration of machine learning guidance with structural insights and sophisticated assay technologies has created a more efficient path from chemical space exploration to validated hits.
Several key principles emerge from these studies:
- Crystal environment matters: For molecular properties influenced by solid-state arrangement, crystal structure prediction must be incorporated into evolutionary algorithms [35]
- Kinetics complement affinity: Binding and signaling kinetics provide crucial insights beyond equilibrium affinity measurements [40] [39]
- Multi-target profiling is feasible: Machine learning approaches enable screening against multiple targets to identify compounds with tailored polypharmacology [32]
- Experimental efficiency is crucial: Multiplex assays that simultaneously monitor multiple pathways in the same well provide more comprehensive pharmacological profiling while conserving resources [39]
As chemical libraries continue to expand toward trillions of compounds, and as structural coverage of the human proteome improves through experimental determination and computational prediction, the active learning approaches described in this case study will become increasingly essential for efficient drug discovery. The integration of these technologies creates a virtuous cycle wherein each successfully discovered compound enhances the predictive models for future campaigns, progressively illuminating the vast darkness of chemical space with the guiding light of computational intelligence.
The exploration of chemical space for functional materials and optimized synthetic protocols represents a frontier as vast and impactful as drug discovery. Framed within the broader thesis of active learning chemical space exploration, this technical guide details how data-driven and physics-informed methodologies are accelerating the development of organic molecular crystals and efficient chemical reactions. We present quantitative benchmarks, detailed experimental protocols, and visual workflows for key computational and experimental strategies, including active learning for identifying complementary reaction conditions, evolutionary algorithms informed by crystal structure prediction, and highly parallel Bayesian optimization. This in-depth review provides researchers with the tools and frameworks necessary to navigate the complexity of chemical and materials space beyond biological applications.
The number of possible small organic molecules is estimated to be on the order of 10^60, presenting a nearly infinite resource for discovering novel functional materials and synthetic routes [13]. However, this vastness also poses a significant challenge; exhaustively searching this space for molecules with desirable properties or optimal reaction conditions is prohibitively expensive using traditional trial-and-error or one-factor-at-a-time (OFAT) approaches [35] [44]. The field is consequently undergoing a paradigm shift, moving from intuition-driven experimentation to computational-led campaigns that strategically guide which experiments to perform.
Central to this shift is the concept of active learning chemical space exploration. In this framework, machine learning models iteratively select the most informative experiments to run based on previous results, effectively balancing the exploration of unknown regions of chemical space with the exploitation of promising areas [45] [46]. This closed-loop approach has proven capable of rapidly identifying high-performance reaction conditions [44] and materials [35] with minimal experimental effort. This guide delves into the application of these advanced computational strategies for two key objectives: optimizing sets of chemical reaction conditions and discovering novel organic molecular materials.
Active Learning for High-Coverage Reaction Condition Sets
A significant challenge in synthetic chemistry is that no single set of reaction conditions can achieve high yields across diverse reactant scaffolds. A powerful solution is to identify small sets of complementary reaction conditions that, when combined, cover a larger region of chemical space than any single condition [45] [47].
Core Methodology and Workflow
The active learning process for discovering these sets involves framing the problem as an iterative coverage optimization task. The algorithm begins with an initial, often small, set of experimental data and a large library of potential reaction conditions. A model is trained to predict the success or yield of reactions under different conditions. The active learning algorithm then selects the next batch of conditions to test experimentally based on their potential to expand the overall coverage of reactant space, often by prioritizing conditions that perform well on reactants which are not yet covered by any high-yielding condition in the current set. The new experimental results are used to update the model, and the loop repeats until a satisfactory set of complementary conditions is identified [45].
Table 1: Key Computational Components for Active Learning of Reaction Conditions
| Component | Description | Example Algorithms/Functions |
|---|---|---|
| Search Space | The defined universe of plausible reaction parameters (catalysts, solvents, additives, temperatures, etc.). | Discrete combinatorial set of 88,000+ conditions [44] |
| Proxy Model | A machine learning model trained to predict reaction outcome (e.g., yield) based on condition parameters. | Gaussian Process (GP) Regressor [44] |
| Acquisition Function | A function that scores all unevaluated conditions to select the most "informative" ones for the next experiment. | Expected Hypervolume Improvement [44] |
| Coverage Metric | The measure of how much of the reactant space is successfully covered by a set of conditions. | Fraction of reactants with yield > threshold [45] |
Experimental Protocol & Key Reagents
A representative experimental validation of this approach involves running a high-throughput experimentation (HTE) campaign. For instance, a Nickel-catalyzed Suzuki reaction can be optimized using a 96-well HTE plate, exploring a search space of over 88,000 possible conditions [44].
Detailed Protocol:
- Reaction Setup: In an automated glovebox, distribute stock solutions of the reactant, base, and Ni-based catalyst precursor to a 96-well plate. A diverse library of ligands and solvents is added according to the condition set selected by the active learning algorithm.
- Execution: The plate is sealed, removed from the glovebox, and heated with agitation on a parallel reactor system.
- Analysis: After quenching, the reaction mixtures are analyzed using ultra-high-performance liquid chromatography with mass spectrometry (UHPLC-MS) to determine yield and selectivity (Area Percent, AP).
- Iteration: The resulting yield/selectivity data is fed back into the active learning algorithm, which selects the next batch of 96 conditions for testing.
Table 2: Research Reagent Solutions for HTE Reaction Optimization
| Reagent Category | Specific Example | Function | Considerations |
|---|---|---|---|
| Catalyst Precursor | Ni(II) salts (e.g., NiCl₂·glyme) | Forms the active catalytic species for cross-coupling. | Earth-abundant and lower-cost alternative to Pd [44]. |
| Ligand Library | Diverse phosphine (e.g., XPhos) and nitrogen-based ligands | Modulates catalyst activity and selectivity. | Critical categorical variable; large diversity is key [44] [48]. |
| Solvent Library | Polar aprotic (DMAc), ethers (1,4-dioxane), aromatics (Toluene) | Dissolves reactants and influences reaction pathway. | Must be selected considering safety and green chemistry guidelines [44] [48]. |
| Base | Inorganic (K₃PO₄) or organic bases | Scavenges protons and facilitates transmetalation step. | Solubility can be a limiting factor. |
Crystal Structure-Aware Evolutionary Algorithms for Materials Discovery
For molecular materials, the target property (e.g., charge carrier mobility) depends not only on the molecule itself but also on its packing in the solid state. Evolutionary algorithms (EAs) that incorporate crystal structure prediction (CSP) directly into the fitness evaluation are a breakthrough for navigating this complexity [35].
Core Methodology and Workflow
The CSP-informed EA (CSP-EA) starts with an initial population of molecules. For each molecule in the population, an automated CSP calculation is performed to generate and rank its likely crystal packing possibilities. The fitness of the molecule (e.g., its predicted charge carrier mobility) is then evaluated based on the properties of its most stable predicted crystal structure(s). The fittest molecules are selected to "reproduce," generating new candidate molecules for the next generation through mutation and crossover operations. This process repeats, with CSP guiding the search toward molecules that are intrinsically predisposed to form crystal structures with the desired properties [35].
Efficient CSP Sampling Protocol
Performing comprehensive CSP for thousands of molecules in an EA is computationally prohibitive. Efficient, coarse sampling schemes are therefore critical. A benchmark study on 20 molecules compared the cost and effectiveness of various schemes against a comprehensive reference search (250,000 structures per molecule) [35].
Detailed CSP Protocol:
- Input: A line notation for the molecule (e.g., InChi string).
- Structure Generation: A low-discrepancy, quasi-random sampling of structural degrees of freedom (unit cell parameters, molecular orientation, and space group) is performed.
- Lattice Energy Minimization: The generated trial crystal structures are optimized using a force field to find local minima on the lattice energy surface.
- Analysis: The lowest energy structures and those within a relevant energy window (e.g., 7.2 kJ mol⁻¹) are identified for property evaluation.
Table 3: Benchmarking of CSP Sampling Schemes for Evolutionary Algorithms
| Sampling Scheme | Space Groups | Structures per Group | Cost (Core-Hours/Molecule) | Low-Energy Structures Recovered |
|---|---|---|---|---|
| Comprehensive (Reference) | 25 | 10,000 | ~2,533 | 100% (by definition) |
| SG14-2000 | 1 (P2₁/c only) | 2,000 | <5 | 33.9% |
| Sampling A | 5 (biased by frequency) | 2,000 | ~76 | 73.4% |
| Top10-2000 | 10 (most frequent) | 2,000 | ~169 | 77.1% |
The data shows that biased sampling of just 5 space groups (Sampling A) recovers nearly three-quarters of the low-energy structures at less than 3% of the computational cost of a comprehensive search, making it highly suitable for EAs [35].
Integrated Frameworks for Scalable Optimization
The Minerva Framework for Highly Parallel HTE
To fully leverage modern automation, optimization frameworks must handle large parallel batches. The Minerva framework combines Bayesian optimization with 96-well HTE, using scalable acquisition functions like q-NParEgo and Thompson Sampling to navigate high-dimensional spaces [44]. In one application, it identified conditions for a Ni-catalyzed Suzuki reaction with 76% AP yield and 92% selectivity, where traditional chemist-designed plates had failed. In pharmaceutical process development, this approach identified conditions with >95% yield and selectivity for API syntheses in weeks instead of months [44].
QUARC for Quantitative Condition Recommendation
Bridging the gap between retrosynthetic planning and experimental execution, the QUARC model provides fully specified, data-driven condition recommendations [48]. It sequentially predicts agent identities, reaction temperature, reactant equivalences, and agent amounts. This structured output provides actionable, executable procedures that serve as excellent warm-starts for active learning optimization campaigns, significantly accelerating the initial experimental phase.
Emerging Frontiers: Foundation Models for Chemical Exploration
Scientific foundation models (SciFMs) represent a paradigm shift by learning generalizable representations of molecular structure from vast, unlabeled datasets. Models like MIST (Molecular Insight SMILES Transformers), with up to 1.8 billion parameters, are trained on billions of molecules using a tokenization scheme that captures nuclear, electronic, and geometric features [13]. After pre-training, MIST can be fine-tuned on hundreds of downstream property prediction tasks, matching or exceeding state-of-the-art performance across diverse domains from electrochemistry to olfaction. This demonstrates a single model's potential to become a universal tool for exploration across chemical space, capable of predicting properties for novel molecules far outside the scope of traditional labeled training sets [13].
The strategic application of active learning, crystal structure prediction, and highly parallel optimization is transforming the exploration of chemical space for materials and reactions. The methodologies and protocols detailed in this guide provide a robust framework for researchers to efficiently discover functional organic molecular crystals and identify high-performing, complementary sets of reaction conditions. As these computational approaches continue to mature and integrate with emerging technologies like scientific foundation models, they promise to significantly accelerate the design and development of novel materials and sustainable chemical processes.
Overcoming Practical Hurdles: Data Scarcity, Generalization, and Optimization
In numerous scientific domains, particularly in chemical and drug discovery research, the scarcity of reliable, high-quality experimental data presents a fundamental obstacle to innovation. Traditional machine learning approaches, which are notoriously "data-hungry," often fail to deliver robust predictions when trained on limited datasets, which is a common scenario for emerging next-generation chemistries [29] [49]. The challenge is compounded by the fact that acquiring each experimental data point can require weeks or even months, making the collection of millions of data points—as often required by conventional models—simply infeasible within practical research timelines [29]. This data bottleneck significantly impedes the pace of materials discovery and drug development, necessitating specialized strategies that can extract maximum insight from minimal data.
The concept of the "low-data regime" is particularly relevant when exploring massive chemical spaces, which can encompass up to 10^60 potential molecules [29]. Within this vast possibility space, researchers must identify promising candidates with only sparse experimental guidance. Framed within the broader context of active learning chemical space exploration research, this whitepaper synthesizes cutting-edge methodologies that enable researchers to thrive within these constraints, transforming data scarcity from a roadblock into a strategic advantage through intelligent experimental design and algorithmic innovation.
Core Strategies for Low-Data Success
Active Learning for Chemical Space Exploration
Active learning represents a paradigm shift from passive data collection to intelligent, adaptive experimentation. This approach employs an iterative cycle where a machine learning model selectively identifies the most informative data points for experimental validation, thereby maximizing the knowledge gained from each experiment [50]. In practice, an initial model is trained on a small starting dataset, then used to predict outcomes across a broader chemical space while simultaneously quantifying prediction uncertainty. The most uncertain or strategically valuable points are prioritized for experimental testing, with results fed back into the model for continuous refinement [29] [50].
This strategy was successfully demonstrated in electrolyte solvent screening for anode-free lithium metal batteries, where researchers started with just 58 initial data points yet efficiently explored a virtual search space of one million potential electrolytes [29]. Through seven active learning campaigns, each testing approximately 10 electrolytes, the team identified four novel electrolytes rivaling state-of-the-art performance. This approach reduced the experimental burden by several orders of magnitude compared to exhaustive screening [29]. Similarly, in predicting ionization efficiency for mass spectrometry, active learning reduced root mean square error (RMSE) by up to 0.3 log units after just a single iteration, significantly improving quantification accuracy from a fold error of 4.13× to 2.94× for natural products in Alpinia officinarum [50].
Multi-Task Learning with Negative Transfer Mitigation
Multi-task learning (MTL) addresses data scarcity by leveraging correlations across related molecular properties or tasks. By sharing representations across tasks, MTL enables models to discover and utilize underlying common structures, thereby improving prediction accuracy for all tasks [51]. However, conventional MTL is often undermined by negative transfer—performance degradation that occurs when updates beneficial for one task are detrimental to another [51]. Negative transfer arises particularly under task imbalance, where certain tasks have far fewer labeled examples than others, a common scenario in real-world research settings [51].
The Adaptive Checkpointing with Specialization (ACS) training scheme effectively mitigates negative transfer while preserving MTL benefits [51]. ACS integrates a shared, task-agnostic backbone (typically a graph neural network) with task-specific trainable heads. During training, the validation loss of every task is monitored, and the best backbone-head pair is checkpointed whenever a task reaches a new validation minimum [51]. This approach enables each task to ultimately obtain a specialized model while still benefiting from shared representations during training. On molecular property prediction benchmarks, ACS consistently surpassed or matched recent supervised methods, demonstrating an 11.5% average improvement relative to other node-centric message passing methods and outperforming single-task learning by 8.3% on average [51]. Notably, ACS enabled accurate predictions with as few as 29 labeled samples in sustainable aviation fuel property prediction—capabilities unattainable with single-task learning or conventional MTL [51].
Data-Efficient Model Architectures
Specialized model architectures can significantly enhance performance in low-data regimes. Graph neural networks (GNNs) have proven particularly effective for molecular property prediction as they naturally represent molecular structure through atoms (nodes) and bonds (edges) [51]. The message-passing mechanism in GNNs enables learning of hierarchical representations that capture both local chemical environments and global molecular properties [51]. This architectural inductive bias reduces the data requirements compared to less structured approaches.
For ionization efficiency prediction, extreme gradient boosting (xgBoost) models trained on PaDEL descriptors have shown strong performance with limited data [50]. The key to success with any architecture lies in proper descriptor management, including cleaning and scaling features, removing near-zero variance descriptors, and eliminating highly correlated features to reduce redundancy and prevent overfitting [50].
Quantitative Performance Comparison
Table 1: Performance Comparison of Low-Data Learning Approaches
| Method | Data Requirements | Performance Highlights | Application Examples |
|---|---|---|---|
| Active Learning | Started with 58 data points, explored 1M candidates [29] | Identified 4 high-performing electrolytes; Reduced IE prediction RMSE by 0.3 log units [29] [50] | Battery electrolyte screening; Ionization efficiency prediction [29] [50] |
| ACS (Multi-Task Learning) | As few as 29 labeled samples [51] | 11.5% average improvement vs. node-centric message passing; 8.3% improvement vs. single-task learning [51] | Molecular property prediction; Sustainable aviation fuel design [51] |
| Single-Task Learning | Substantially higher requirements [51] | Underperformed ACS by 8.3% on average [51] | Baseline comparison in molecular property benchmarks [51] |
| Conventional MTL | Similar to ACS but less efficient [51] | Outperformed single-task by 3.9% but trailed ACS [51] | Molecular property prediction with negative transfer issues [51] |
Table 2: Active Learning Sampling Strategies Comparison
| Sampling Approach | Key Mechanism | Efficiency Notes | Best Use Cases |
|---|---|---|---|
| Uncertainty-Based | Selects points with highest prediction uncertainty [50] | Inefficient if ≥10 chemicals sampled per iteration [50] | Rapid initial improvement with very low sampling numbers [50] |
| Clustering-Based | Chooses representatives from different clusters [50] | Reduced RMSE least in IE prediction studies [50] | Ensuring broad coverage of chemical space [50] |
| Anti-Clustering | Selects diverse candidates across distribution [50] | Not the most effective standalone approach [50] | Complementary strategy to balance exploration [50] |
| Mixed Algorithm | Combines clustering and uncertainty [50] | Balances exploration and uncertainty reduction [50] | Practical applications requiring robust performance [50] |
| Random Sampling | Baseline random selection [50] | Inefficient compared to directed approaches [50] | Control experiments and baseline establishment [50] |
Experimental Protocols and Methodologies
Active Learning Implementation Protocol
Implementing an effective active learning cycle requires careful design at each stage. The following protocol outlines a robust methodology validated in electrolyte discovery and ionization efficiency prediction studies [29] [50]:
Initial Model Training: Begin with a small but diverse starting set (as few as 58 data points can suffice). Train an initial model using appropriate descriptors (e.g., PaDEL descriptors for molecules) and architecture (e.g., graph neural networks for molecular properties) [29] [50].
Chemical Space Representation: Map the unexplored chemical space using dimensionality reduction techniques like uniform manifold approximation and projection (UMAP). Calculate similarity as Euclidean distance from chemicals in the unexplored space to the nearest neighbors in the explored space based on scaled descriptors [50].
Informativeness Quantification: Deploy multiple sampling strategies in parallel or sequence:
- Compute prediction uncertainties for all candidates
- Identify diverse candidates through clustering
- Balance exploration vs. exploitation based on campaign progress [50]
Experimental Validation: Prioritize the most informative candidates for experimental testing. In battery electrolyte studies, this involved actually building batteries with suggested electrolytes and cycling them to obtain performance data [29].
Iterative Model Refinement: Incorporate new experimental results into the training set and retrain the model. Implement early stopping based on validation performance to prevent overfitting [29] [50].
Termination Criteria: Continue cycles until meeting one of:
- Performance targets achieved
- Experimental budget exhausted
- Diminishing returns observed [29]
ACS Training Scheme Methodology
The Adaptive Checkpointing with Specialization approach requires specific implementation details to achieve optimal performance [51]:
Architecture Setup:
- Implement a shared graph neural network backbone based on message passing
- Attach task-specific multi-layer perceptron heads for each property
- Use loss masking for missing labels to handle task imbalance [51]
Training Procedure:
- Monitor validation loss for every task throughout training
- Checkpoint the best backbone-head pair whenever a task reaches new minimum validation loss
- Employ gradient-based optimization with careful learning rate selection [51]
Task Imbalance Quantification:
- Calculate task imbalance using the formula: Iᵢ = 1 - (Lᵢ / maxⱼ Lⱼ)
- Where Lᵢ is the number of labeled entries for task i [51]
Specialization Phase:
- After training, obtain a specialized model for each task by selecting the checkpointed backbone-head pair that achieved that task's best performance [51]
Workflow Visualization
Active Learning Workflow for Chemical Discovery
ACS Architecture with Adaptive Checkpointing
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Computational Tools
| Resource Category | Specific Examples | Function and Application | Implementation Notes |
|---|---|---|---|
| Molecular Descriptors | PaDEL descriptors [50] | Numerical representation of chemical structures for machine learning | Clean and scale descriptors; remove near-zero variance and highly correlated features [50] |
| Model Architectures | Graph Neural Networks (GNNs) [51]; xgBoost [50] | Learning molecular representations and property prediction | GNNs for structured data; xgBoost for tabular descriptor data [51] [50] |
| Experimental Materials | FCF Brilliant Blue dye [52]; Battery components [29] | Experimental validation of computational predictions | Standardized materials ensure reproducible experimental results [29] [52] |
| Analysis Tools | XLMiner ToolPak (Google Sheets) [52]; Analysis ToolPak (Excel) [52] | Statistical analysis of experimental results | Perform t-tests, F-tests to validate significance of findings [52] |
| Benchmark Datasets | MoleculeNet benchmarks [51]; Sustainable aviation fuel data [51] | Model validation and performance comparison | Includes ClinTox, SIDER, Tox21 for fair comparison [51] |
Thriving in low-data regimes requires a fundamental shift from data-intensive brute force approaches to intelligent, adaptive strategies that maximize information gain from each experiment. Active learning provides a powerful framework for guided exploration of massive chemical spaces, while advanced multi-task learning approaches like ACS leverage correlations across related properties to overcome individual data limitations. These methodologies, combined with appropriate model architectures and careful experimental design, enable researchers to accelerate discovery even when extensive data collection is impractical. As these approaches continue to mature, they promise to democratize research access and accelerate the pace of innovation across chemical discovery, materials science, and drug development.
In the realm of active learning for chemical space exploration, the efficient navigation of vast, multidimensional experimental landscapes is paramount. Acquisition functions serve as the intellectual core of Bayesian optimization (BO), guiding the iterative process of experiment selection by automating the critical trade-off between exploration and exploitation. Exploitation involves selecting experiment conditions predicted to yield high performance based on existing data, while exploration prioritizes sampling in uncertain regions of the chemical space to gather new information and avoid local optima. For researchers and drug development professionals, mastering this balance is not merely a theoretical exercise; it is a practical necessity for accelerating the discovery of functional molecules, optimizing synthetic protocols, and mapping complex structure-property relationships with limited experimental resources.
The challenge is particularly acute in chemistry and drug discovery, where the experimental evaluation of a single candidate—be it a reaction condition, an alloy composition, or a drug molecule—can be prohibitively expensive and time-consuming. Bayesian optimization, powered by sophisticated acquisition functions, has emerged as a powerful framework to address this challenge, enabling autonomous experimentation and the efficient traversal of chemical spaces containing billions of potential candidates [32].
Core Mathematical Principles of the Exploration-Exploitation Trade-Off
An acquisition function, denoted as ( \alpha(x) ), is a function that quantifies the utility of evaluating a candidate point ( x ) in the experimental space (e.g., a set of reaction parameters or a molecular descriptor). The next experiment is chosen by maximizing this function: ( x{next} = \arg\max{x \in X} \alpha(x) ). The mathematical form of ( \alpha(x) ) is designed to balance two competing objectives based on the predictions of a surrogate model, typically a Gaussian Process (GP).
A Gaussian Process provides a probabilistic prediction for any point ( x ), characterized by a mean function ( \mu(x) ) (the expected value of the objective) and a variance function ( \sigma^2(x) ) (the uncertainty around that prediction) [53]. The mean represents the model's best guess, while the variance represents its confidence.
- Exploitation is guided by the mean ( \mu(x) ). A purely exploitative strategy would select ( x = \arg\max \mu(x) ), always trusting the model's current best prediction.
- Exploration is driven by the variance ( \sigma(x) ). A purely exploratory strategy would select ( x = \arg\max \sigma(x) ), probing the region where the model is most uncertain.
The most common acquisition functions combine these two elements. A canonical example is the Upper Confidence Bound (UCB): [ \alpha_{UCB}(x) = \mu(x) + \kappa \sigma(x) ] Here, ( \kappa ) is a tunable parameter that explicitly controls the balance between exploration and exploitation [53]. A higher ( \kappa ) value favors exploration, encouraging the algorithm to investigate uncertain regions. A lower ( \kappa ) prioritizes exploitation, focusing on regions predicted to be high-performing.
Table 1: Common Acquisition Functions and Their Characteristics
| Acquisition Function | Mathematical Formulation | Balance Mechanism | Typical Use Case |
|---|---|---|---|
| Upper Confidence Bound (UCB) | ( \mu(x) + \kappa \sigma(x) ) | Direct parameter ( \kappa ) | Intuitive and widely applicable; good for chemical process optimization [53]. |
| Expected Improvement (EI) | ( \mathbb{E}[\max(f(x) - f(x^+), 0)] ) | Improves over current best ( f(x^+) ) | Focused search for peak performance; common in materials synthesis. |
| Probability of Improvement (PI) | ( P(f(x) \ge f(x^+) + \xi) ) | Trade-off parameter ( \xi ) | Simpler than EI, but can be less efficient. |
Another critical strategy for managing this balance is the integration of known constraints from chemistry. Experimental conditions often have interdependent, non-linear constraints (e.g., temperature and pressure limits, synthetic accessibility). By incorporating these directly into the acquisition function or the optimization domain, algorithms can avoid recommending invalid or dangerous experiments, significantly improving search efficiency and practical utility [54].
Implementation in Chemical Research
The theoretical framework of acquisition functions is brought to life in various chemical research scenarios, demonstrating its transformative potential.
Optimizing Magnesium Alloys
An active machine learning approach using Bayesian optimization was successfully deployed for the optimal design of magnesium (Mg) alloys. The workflow used a Gaussian process regressor as a surrogate model and the UCB acquisition function to balance the exploration of new alloy compositions and the exploitation of known high-performance regions [55]. This approach efficiently navigated a complex dataset of 916 unique Mg alloys, which varied in composition (30 different alloying elements) and thermomechanical processing conditions. The goal was to optimize conflicting mechanical properties like strength and ductility, a classic multi-objective problem where the acquisition function guided the search towards the optimal trade-off. The performance of this sequential strategy was rigorously validated via regret analysis, which quantified the difference between the ideal optimal property value and the value found by the optimizer over successive iterations [55].
Virtual Screening of Ultralarge Chemical Libraries
In drug discovery, a key challenge is virtually screening make-on-demand chemical libraries containing billions of compounds. A machine learning-guided docking screen was developed to address this. A CatBoost classification algorithm was trained to predict top-scoring compounds from molecular docking simulations [32]. The conformal prediction framework was then used as an intelligent filter, selecting a much smaller subset of compounds for explicit docking. This workflow effectively uses an acquisition-like strategy: the classifier exploits known structure-activity relationships, while the conformal predictor explores the vast chemical space by controlling the error rate of predictions. This hybrid approach achieved a 1,000-fold reduction in computational cost, enabling the practical screening of a 3.5-billion-compound library and leading to the experimental identification of ligands for G protein-coupled receptors (GPCRs) [32].
Mapping Post-Stroke Brain Function
While not strictly a chemistry application, a related methodology from neuroscience illustrates the power of intelligent search. Neuroadaptive Bayesian optimization was used to rapidly map patient-specific profiles of residual brain network function post-stroke. The algorithm efficiently searched a large space of cognitive tasks to identify those that maximally activated target brain networks [56]. This mirrors the chemical challenge of searching a high-dimensional space with expensive evaluations (fMRI scans in this case), demonstrating how Bayesian optimization can extract maximum information from a limited number of experiments.
Experimental Protocols and Workflows
Implementing Bayesian optimization with a well-designed acquisition function follows a structured experimental protocol.
General Bayesian Optimization Workflow
The following diagram illustrates the core, iterative feedback loop of a Bayesian optimization experiment.
Title: Bayesian Optimization Feedback Loop
Protocol Steps:
- Initialization: Begin with a small initial dataset ( D0 = {(xi, yi)}{i=1}^n ) of previously conducted experiments or a set chosen by an experimental design method (e.g., D-optimal design) [53].
- Surrogate Model Training: Fit a Gaussian Process (GP) model to the current dataset ( Dt ). The GP provides a probabilistic prediction ( P(y|x, Dt) ) for any point ( x ) in the chemical space, characterized by a mean ( \mu(x) ) and uncertainty ( \sigma(x) ) [53].
- Acquisition Function Maximization: Construct the acquisition function ( \alpha(x) ) using the GP's outputs. The next experiment point ( x{next} ) is selected by solving ( x{next} = \arg\max_{x \in X} \alpha(x) ). This is the step where the exploration-exploitation balance is enacted [53].
- Experiment Evaluation: Conduct the wet-lab experiment, synthesis, or simulation at the proposed condition ( x{next} ) to obtain the result ( y{next} ).
- Data Update: Augment the dataset with the new result: ( D{t+1} = Dt \cup {(x{next}, y{next})} ).
- Iteration: Repeat steps 2-5 until a stopping criterion is met, such as convergence, a maximum number of experiments, or the achievement of a target performance threshold.
Integrated BODO Workflow for Robust Performance
To mitigate BO's known sensitivity to poor initial data, the BODO (Bayesian Optimization with D-optimal Design) framework introduces a stochastic branching step that enhances exploration [53].
Title: BODO Stochastic Branching Workflow
Key Modifications to the Base Protocol:
- Stochastic Branching: After training the GP, the algorithm randomly selects between two branches with a defined probability.
- BO Branch (Exploitation): This branch uses a standard acquisition function like UCB to propose a candidate, focusing on predicted high performance.
- D-optimal Branch (Exploration): This branch proposes a candidate that maximizes the D-optimality criterion, which seeks points that maximize the information content about the model parameters, often favoring regions with less data [53].
- Final Selection: The final candidate from the chosen branch is evaluated based on its D-optimal criterion value, ensuring the selected experiment is highly informative regardless of its origin.
The Scientist's Toolkit: Research Reagents & Computational Solutions
The application of these advanced optimization strategies requires a suite of computational "reagents" and tools.
Table 2: Key Computational Tools for Active Learning in Chemistry
| Tool / Solution | Function | Relevance to Exploration/Exploitation |
|---|---|---|
| Gaussian Process (GP) Regressor | Probabilistic surrogate model that provides predictions with uncertainty estimates. | The backbone of BO; the uncertainty quantification (( \sigma )) is the primary driver for exploration [55] [53]. |
| PHOENICS & GRYFFIN | Bayesian optimization algorithms specifically designed for chemical applications. | These tools have been extended to handle arbitrary known experimental constraints, making the exploration of complex chemical spaces safer and more efficient [54]. |
| CatBoost Classifier | A high-performance gradient-boosting algorithm for classification tasks. | Used for rapid pre-screening in virtual screening; its speed allows for broad exploration, while its accuracy enables effective exploitation of predicted actives [32]. |
| Conformal Prediction (CP) Framework | A method for generating predictions with guaranteed confidence levels. | Provides a statistically rigorous way to define and control the exploration of a chemical library by managing the error rate in candidate selection [32]. |
| D-Optimal Design | A criterion from the Design of Experiments (DoE) for selecting informative points. | Integrated into frameworks like BODO to force exploration of the independent variable space, reducing the risk of becoming stuck in local optima [53]. |
The strategic balancing of exploration and exploitation through acquisition functions is a cornerstone of modern, data-driven chemical research. As laboratory automation and accessible high-performance computing continue to evolve, these algorithms will become increasingly integral to the scientist's workflow. Frameworks that intelligently manage this balance—such as constrained BO for complex experimental setups and hybrid ML-docking screens for vast chemical libraries—are pushing the boundaries of what is possible in a research timeline. The future of chemical discovery lies in the seamless integration of these computational strategies with automated experimental platforms, creating a closed-loop system where algorithms not only suggest the most informative next experiment but also execute them, dramatically accelerating the pace of scientific discovery.
The exploration of chemical space for novel drug candidates is a monumental challenge, with the number of potential molecules estimated to be as high as 10^60 [29]. Traditional experimental approaches are often impractical due to the prohibitive time and cost required to synthesize and test even a tiny fraction of this space. Within this context, active learning has emerged as a transformative paradigm, enabling efficient navigation of chemical space by iteratively guiding experiments through intelligent, data-driven selection. This guide details the integration of active learning methodologies to simultaneously optimize the complex multi-objective landscape defined by synthesizability, ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity), and potency—a trinity of properties essential for successful drug development.
This technical framework is designed for researchers and drug development professionals. It moves beyond theoretical discussion to provide actionable protocols, quantitative performance data, and visual workflows to implement these strategies in practical research settings, from initial setup to final validation.
Active Learning for Chemical Space Exploration
Active learning (AL) is a machine learning subfield that addresses scenarios where labeled data is scarce or expensive to acquire, as is the case with experimental chemistry and biology. Its core function is to identify the most informative data points from a vast unexplored space to be measured experimentally, thereby maximizing model performance with a minimal labeling budget [50].
Core Workflow and Sampling Strategies
The AL cycle is an iterative process that closes the loop between computational prediction and experimental validation. The workflow begins with an initial, often small, training set. A machine learning model is trained on this data and used to predict the properties of a larger, unlabeled chemical pool. The key step is using a sampling algorithm to select the most promising candidates from this pool for experimental testing. The results of these experiments are then fed back into the training set, and the model is retrained, progressively enhancing its accuracy and guiding the exploration toward regions of chemical space that meet the desired objectives [29] [50].
The choice of sampling strategy is critical for efficient exploration. The table below summarizes the primary approaches used in active learning for chemical research.
Table 1: Common Active Learning Sampling Strategies for Chemical Space Exploration
| Strategy | Mechanism | Advantage | Disadvantage |
|---|---|---|---|
| Uncertainty-Based | Selects chemicals for which the model's prediction is most uncertain. | Highly efficient at improving model accuracy for specific predictions. | Can be inefficient if many chemicals are sampled in one iteration; may explore local regions. [50] |
| Clustering-Based | Selects chemicals that are geographically diverse in the chemical descriptor space. | Broadly expands the chemical space coverage of the training set. | May include data points that do not significantly improve model accuracy for a targeted space. [50] |
| Anti-Clustering | Aims to select a set of chemicals that are as similar as possible to the overall distribution of the unexplored space. | Promotes diversity and representativeness in the selected batch. | Computational cost can be higher than simple clustering. [50] |
| Mixed (Hybrid) | Combines multiple approaches, e.g., clustering first, then selecting the most uncertain from each cluster. | Balances exploration (diversity) with exploitation (uncertainty). | Requires tuning of the balance between the different selection criteria. [50] |
The following diagram illustrates the iterative workflow of an active learning cycle, integrating these sampling strategies with experimental validation.
Figure 1: The Active Learning Cycle for Drug Discovery. This iterative process closes the loop between computational prediction and experimental validation to efficiently optimize multiple objectives.
Quantitative Performance of Active Learning
Recent studies demonstrate the profound impact of active learning on accelerating materials and drug discovery. A landmark study on battery electrolyte development showcased the power of this approach, where an active learning model, starting from only 58 initial data points, explored a virtual search space of one million potential electrolytes. Through seven iterative campaigns, each testing about ten electrolytes, the model successfully identified four distinct new electrolyte solvents that rivaled state-of-the-art performance [29]. This represents an experimental efficiency improvement of several orders of magnitude.
In a more direct drug discovery context, research on predicting ionization efficiency (IE) for mass spectrometry quantification compared active learning strategies. The study found that the root mean square error (RMSE) in the targeted chemical space "dropped significantly (up to 0.3 log units) after a single AL iteration" [50]. The table below, adapted from this research, shows how expanding the chemical space with active learning improved quantification accuracy for natural products.
Table 2: Impact of Active Learning on Ionization Efficiency (IE) Prediction and Quantification Accuracy for Natural Products [50]
| Chemical | Reference Concentration (µM) | Fold Error (Before AL) | Fold Error (After AL) |
|---|---|---|---|
| Pinocembrin | 15 | 4.13 | 2.94 |
| (-)-Epicatechin | 23 | 3.39 | 1.78 |
| Kaempferol | 1.04 | 1.15 | 2.31 |
| Galangin | 110 | 1.67 | 1.33 |
| Luteolin-3',7-diglucoside | 0.16 | 2.26 | 1.08 |
| Average Fold Error | 2.52 | 1.89 |
The data shows that active learning consistently improved quantification accuracy for most compounds, reducing the average fold error. This principle translates directly to multi-objective optimization, where more accurate predictive models lead to more reliable candidate selection.
A Multi-Objective Optimization Framework
The ultimate goal is to find molecules that are not only potent but also synthesizable and possess favorable ADMET profiles. This requires a framework that can balance these competing objectives.
Defining the Objectives
- Potency: Typically measured by IC50, EC50, or Ki in experimental assays. The primary driver for initial candidate selection.
- ADMET: A composite of critical pharmacokinetic and safety properties. Key metrics include membrane permeability (e.g., Caco-2 assay), metabolic stability (e.g., human liver microsome half-life), and potential for drug-drug interactions (e.g., CYP inhibition).
- Synthesizability: A predictive measure of the ease with which a molecule can be synthesized. This can be assessed using computational scores (e.g., SAscore, SCScore) or by evaluating the number of synthetic steps, the commercial availability of starting materials, and the complexity of required reactions.
Integrated Workflow and Experimental Design
Success in multi-objective optimization hinges on a structured workflow that integrates computational and experimental layers. The process begins with defining the chemical space and acquiring initial data, often from public databases or legacy in-house data. The next critical step is featurization, representing molecules using numerical descriptors (e.g., PaDEL descriptors, fingerprints, or topological indices) [50]. A multi-task machine learning model is then trained to predict all key objectives—potency, ADMET properties, and synthesizability—simultaneously.
An active learning loop is initiated from this point. The model's predictions are used to score and rank compounds in the unexplored chemical space. A multi-objective selection criterion, such as Pareto optimization or a weighted scoring function, is applied to identify the most promising candidates that balance all desired properties. These candidates are then synthesized and tested in the appropriate assays, and the results are used to update the model, continuing the cycle.
The statistical design of experiments (DoE) is highly recommended for structuring this validation phase. Techniques like Response Surface Methodology (RSM) can systematically optimize experimental conditions and evaluate the influence of multiple factors on the outcomes, ensuring robust and interpretable results [57].
Experimental Protocols and Data Standards
Reproducibility and data quality are the bedrock of reliable active learning. Adhering to community standards is paramount.
Protocol for a High-Throughput Screening Cycle
This protocol outlines a single iteration of the active learning loop for multi-objective optimization.
Computational Candidate Selection:
- Input: A library of molecular structures (e.g., >1M compounds) and a model trained on initial data.
- Procedure: Use the trained multi-task model to predict potency, ADMET endpoints, and synthesizability for all compounds in the library. Apply a multi-objective filter (e.g., Pareto front selection based on predicted potency, synthesizability score, and low predicted CYP3A4 inhibition) to select a shortlist of 50-200 candidates.
- Output: A list of candidate molecules for synthesis.
Chemical Synthesis and Characterization:
- Procedure: Synthesize the selected candidates. Adhere to rigorous analytical standards.
- Data Reporting: For all key compounds, report
1H NMR, 13C NMR, and HRMSdata. The purity and method of purity determination (e.g., analytical HPLC) must be reported [58].
In Vitro Potency and ADMET Assay:
- Cell-Based Potency Assay: Conduct a dose-response assay to determine the half-maximal inhibitory concentration (IC50). Run in triplicate with appropriate positive and negative controls.
- Microsomal Stability Assay: Incubate compounds (1 µM) with human liver microsomes. Measure parent compound depletion over 45 minutes to determine half-life and calculate intrinsic clearance.
- Caco-2 Permeability Assay: Assess bidirectional transport to determine apparent permeability (Papp) and efflux ratio.
- Data Reporting: Clearly state the source of biological materials (e.g., cell lines, microsomes), assay conditions, and a description of the statistical analysis. All experiments must include justification for the number of replicates using a power analysis where applicable [58].
Model Retraining:
- Procedure: Integrate the new experimental data (synthesis success/failure, IC50, clearance, Papp) into the training set. Clean the updated data set by removing descriptors with near-zero variance or high correlation (>0.75) to reduce redundancy [50]. Retrain the multi-task model with the updated dataset.
Data and Reporting Guidelines
To ensure reproducibility and build high-quality datasets, follow these guidelines:
- Data Deposition: Deposit data in discipline-specific, community-recognized repositories that issue persistent identifiers (e.g., DOI). For biological sequence data, use repositories like GenBank or GEO [58].
- Analytical Figures of Merit: Report data on reproducibility, accuracy, selectivity, sensitivity, and detection limits to allow others to assess the performance of the method [58].
- Biological Reagents: For antibodies, report the name, host species, catalog number, and application. For cell lines, report the source, date of authentication, and method of authentication [58].
- Statistical Reporting: In figure and table captions, include the number of replicates, the method of statistical analysis, and the corresponding p-values for significant differences [58].
The Scientist's Toolkit
Implementing an active learning-driven discovery pipeline requires a suite of computational and experimental tools.
Table 3: Research Reagent Solutions for Active Learning in Drug Discovery
| Tool / Reagent | Function | Application in Workflow |
|---|---|---|
| PaDEL Descriptors | Software to compute molecular descriptors and fingerprints for chemical structures. | Featurization of molecules for machine learning model training [50]. |
| XGBoost | A scalable and efficient machine learning algorithm based on gradient boosting. | Building predictive models for potency, ADMET, and synthesizability [50]. |
| Human Liver Microsomes | Subcellular fraction containing drug-metabolizing enzymes (CYPs, UGTs). | Experimental assessment of metabolic stability in ADMET assays [58]. |
| Caco-2 Cell Line | A human colon adenocarcinoma cell line that differentiates into enterocyte-like cells. | In vitro model for predicting intestinal permeability and efflux [58]. |
| Analytical HPLC-MS | High-Performance Liquid Chromatography coupled with Mass Spectrometry. | Determining compound purity and characterizing chemical structures during synthesis [58]. |
| Response Surface Methodology (RSM) | A collection of statistical techniques for designing experiments, building models, and optimizing processes. | Systematically optimizing experimental conditions and understanding factor interactions [57]. |
The integration of active learning into the drug discovery pipeline represents a paradigm shift from sequential, high-throughput screening to an intelligent, iterative process of hypothesis generation and testing. By framing the challenge as a multi-objective optimization problem—balancing synthesizability, ADMET, and potency—researchers can direct precious experimental resources toward the most promising regions of chemical space. The structured workflows, experimental protocols, and toolkits outlined in this guide provide a concrete foundation for research teams to implement these strategies. As these methodologies mature, they hold the promise of significantly accelerating the discovery of viable drug candidates, reducing late-stage attrition, and ultimately delivering new medicines to patients more efficiently.
The integration of Human-in-the-Loop (HITL) methodologies with Reinforcement Learning from Human Feedback (RLHF) represents a paradigm shift in computational scientific discovery, particularly within chemical space exploration. While traditional machine learning approaches operate autonomously once trained, they often fail to capture the nuanced, tacit knowledge that domain experts accumulate through years of experience. This knowledge gap becomes critically apparent in drug discovery, where molecular suitability encompasses not merely binding affinity but synthesizability, toxicity profiles, metabolic stability, and other pharmacologically relevant properties that often elude purely computational metrics.
The RLHF framework, originally developed for aligning large language models with human values [59] [60], offers a structured approach to embedding expert knowledge directly into the optimization loop. In scientific domains, this translates to creating continuous feedback cycles where computational models propose candidates, experts evaluate them based on multifaceted criteria, and reward models iteratively refine their understanding of what constitutes a "high-quality" suggestion. This whitepaper examines the technical implementation, experimental protocols, and quantitative benefits of integrating expert feedback with RLHF, with specific application to active learning frameworks for navigating chemical space in drug development.
Core Principles: RLHF and Active Learning
The RLHF Framework
Reinforcement Learning from Human Feedback is a multi-stage process designed to align machine learning outputs with complex, human-defined preferences that are difficult to codify into explicit reward functions [59] [60]. The standard workflow, which can be adapted for scientific discovery, consists of three primary phases:
- Phase 1: Supervised Fine-Tuning (SFT) - A base model (e.g., a molecular generator) is initially fine-tuned on a high-quality, expert-curated dataset. This provides the model with foundational knowledge about the domain, such as chemical synthetic rules or basic structure-activity relationships.
- Phase 2: Reward Model Training - The core of RLHF involves training a separate reward model to predict expert preferences. Multiple outputs (e.g., generated molecules) for the same input are presented to domain experts who rank them from best to worst. These rankings form a preference dataset used to train the reward model to output a scalar reward score that mimics human judgment [59].
- Phase 3: Reinforcement Learning Optimization - The base model is optimized using RL algorithms (e.g., Proximal Policy Optimization) with the learned reward model as the objective function. The model's policy is adjusted to maximize the expected reward, thereby aligning its outputs with expert preferences without further direct human intervention [61].
Active Learning in Chemical Space Exploration
Active learning addresses a fundamental challenge in drug discovery: the prohibitive cost of obtaining high-fidelity experimental data for vast chemical libraries. It operates through an iterative cycle of sampling, evaluation, and model updating [62]. In this paradigm:
- A surrogate model provides rapid, inexpensive but approximate predictions across the chemical space.
- An oracle (traditionally wet-lab experiments or high-precision simulations) provides ground-truth validation for a strategically selected subset of candidates.
- An acquisition function determines which candidates are most valuable to evaluate next, typically balancing exploration (sampling uncertain regions) and exploitation (sampling predicted high-performers) [63].
The fusion of RLHF with active learning creates a powerful synergy. The reward model in RLHF can function as an enhanced surrogate, trained not on a simple physicochemical property but on a composite reward reflecting expert holistic judgment. Meanwhile, the domain expert transitions from a passive data provider to an active feedback mechanism within the optimization loop.
Integrated Framework: Expert-Centric RLHF for Chemical Discovery
The proposed integrated framework, which we term Expert-RLHF, consists of a structured workflow where computational models and human expertise interact iteratively. The diagram below illustrates this process and its key decision points.
Figure 1: Expert-RLHF Integrated Workflow for Chemical Discovery
Workflow Description
The Expert-RLHF framework transforms the traditional active learning loop by embedding a learned reward model trained on expert preferences:
- Initialization: The process begins with a base molecular generator (e.g., a graph neural network) that is supervisedly fine-tuned on a dataset of known bioactive compounds or synthetic pathways to establish basic competence [64].
- Candidate Generation: The fine-tuned model generates a diverse set of candidate molecules.
- Expert Evaluation: Domain experts (medicinal chemists, pharmacologists) evaluate and rank these candidates based on multi-factorial criteria including synthetic feasibility, novelty, and perceived drug-likeness that go beyond simple quantitative estimates.
- Reward Model Training: The expert rankings train a reward model to predict a composite "expert preference score."
- RL Optimization: The generator is optimized via reinforcement learning against the frozen reward model, aligning its output distribution with expert preferences.
- Iteration and Validation: The cycle repeats, with the reward model periodically updated with fresh expert feedback. High-scoring candidates from the final cycle are forwarded for experimental validation.
Experimental Protocols & Methodologies
Case Study: SynGFN with Integrated Expert Feedback
Recent advancements in generative models for synthesizable molecules provide a foundation for implementing Expert-RLHF. The SynGFN (Synthetic Generative Flow Network) framework demonstrates how to incorporate synthetic constraints directly into the generation process [64]. The protocol below outlines how to augment such a system with RLHF.
Objective: To discover novel, synthesizable inhibitors for a specified protein target (e.g., GluN1/GluN3A) with desirable drug-like properties. Base Model: SynGFN, which models molecular construction as a Markov decision process under the constraint of known reaction templates, ensuring synthetic feasibility [64].
Step-by-Step Protocol:
Baseline Model Pre-training:
- Train SynGFN on a curated set of drug-like molecules and their synthetic pathways using a building-block library (e.g., Enamine's砌块库) [64].
- Initialize the policy network (π) to sample molecular assembly actions (reaction template + building blocks) with probabilities proportional to a reward based on a target property (e.g., QED).
Initial Generation and Expert Feedback Loop:
- Sample the first generation of 1,000 molecules from the pre-trained SynGFN.
- Present a diverse subset of 100-200 molecules to a panel of 3-5 expert medicinal chemists.
- Experts rank molecules in a paired comparison format (A/B tests) based on multi-dimensional criteria provided in the table below.
- Collect a minimum of 5,000 pairwise comparisons to ensure robust reward model training [65].
Reward Model Training:
- Represent each molecule using its molecular fingerprint and descriptor features.
- Train a neural network reward model (RM) using the Bradley-Terry model on the collected preference data, where the probability that molecule A is preferred over B is given by:
σ(RM(A) - RM(B)), where σ is the logistic function [59].
RL Fine-tuning with Proximal Policy Optimization (PPO):
- Freeze the trained reward model.
- Use the reward model's output as the reward signal for fine-tuning the SynGFN policy network with PPO.
- The objective is to maximize the expected reward:
E_τ~π(τ)[RM(τ)], where τ is a generation trajectory (sequence of actions leading to a molecule) [61].
Iterative Batch Refinement:
- Generate a new batch of molecules with the fine-tuned policy.
- Select molecules for the next expert evaluation round using an acquisition function that balances high reward (exploitation) and high uncertainty from the reward model (exploration).
- Update the reward model with new expert data and repeat steps 4-5 for 3-5 cycles.
Table 1: Key Evaluation Criteria for Expert Feedback in Molecular Design
| Criteria Category | Specific Metrics | Expert Evaluation Guidance |
|---|---|---|
| Synthetic Feasibility | Retrosynthetic complexity, Availability of building blocks, Number of synthetic steps | Rank molecules with clear, short synthetic paths higher. |
| Drug-Likeness | QED, logP, Molecular Weight, Topological Polar Surface Area (TPSA) | Prefer molecules within Lipinski's Rule of 5 and Veber's criteria. |
| Structural Novelty | Tanimoto similarity to known actives, Scaffold uniqueness | Prioritize novel chemotypes that avoid known patent space. |
| Toxicity Risk | Structural alerts, Predicted off-target interactions | Penalize molecules with known toxicophores (e.g., reactive groups). |
Case Study: Multi-Fidelity Active Learning with 3D-QSAR and FEP
Another effective protocol combines RLHF with a multi-fidelity active learning strategy, demonstrating significant resource savings. This approach, as documented in a study combining 3D-QSAR with Free Energy Perturbation (FEP) calculations, uses a surrogate-oracle architecture [63].
Protocol:
Role Definition:
- Surrogate Model (Fast, Approximate): 3D-QSAR models (Consensus and Gaussian Process Regression) provide rapid predictions and uncertainty estimates for large compound libraries [63].
- Oracle (Slow, Accurate): FEP calculations provide near-experimental accuracy for binding affinity prediction but are computationally expensive [63].
Active Learning Loop:
- Initial Phase: Perform FEP calculations on a small, diverse initial set of 30 compounds to generate high-quality training data.
- Surrogate Training: Train the 3D-QSAR models on the FEP data.
- Intelligent Querying:
- The surrogate screens a large virtual library (e.g., 500 compounds).
- Select the next batch for FEP calculation by combining:
- Exploitation: Top 10 predictions from the consensus model.
- Exploration: 20 molecules with the highest uncertainty from the Gaussian Process model.
- Iteration: Update the surrogate model with new FEP results and repeat for 2-3 cycles.
Integration of RLHF:
- Expert chemists can review the selected batches before FEP calculation, ranking or filtering candidates based on synthetic feasibility or other criteria not captured by the affinity prediction.
- These expert preferences train a reward model that can be used to re-rank the selection from the surrogate model, ensuring that compounds advanced to expensive FEP are both predicted to be potent and deemed synthesizable and desirable by experts.
Table 2: Quantitative Performance of Active Learning vs. Traditional Screening
| Screening Method | Computational Cost (FEP Calculations) | Hit Discovery Efficiency | Key Performance Metrics |
|---|---|---|---|
| Traditional Virtual Screening | 500 compounds (100%) | Baseline | Identified 4 known actives in top 20 [63] |
| Active Learning (3D-QSAR + FEP) | 80 compounds (16%) | 2.5x improvement | Identified 10 known actives in top 20 [63] |
| Active Learning on PDE2 Inhibitors | Small subset of library | Robust identification of true positives | Efficient navigation of large chemical library [26] |
| SynGFN Exploration | N/A (Generative Model) | 70x coverage increase | Highest chemical space coverage vs. baselines [64] |
The Scientist's Toolkit: Essential Research Reagents & Solutions
Successful implementation of the Expert-RLHF framework requires a suite of computational and experimental tools. The following table details the key components.
Table 3: Essential Reagents and Tools for Expert-RLHF Experiments
| Tool Category | Specific Tool/Resource | Function in the Workflow |
|---|---|---|
| Generative Models | SynGFN [64], REINVENT [26] | Generates novel molecular structures within synthesizable chemical space. |
| Chemical Libraries | Enamine Building Blocks [64], ZINC, ChEMBL | Provides foundational chemical structures and reactants for virtual library construction and generative model training. |
| Simulation & Oracle | Flare FEP [63], Alchemical Free Energy Calculations [26] | Provides high-accuracy, computationally derived binding affinities to act as ground truth (Oracle). |
| Surrogate Models | 3D-QSAR [63], Random Forest, CNN [66] | Provides fast, approximate property predictions (e.g., toxicity, affinity) for initial screening and uncertainty estimation. |
| Reward Model Training | PyTorch, TensorFlow, Bradley-Terry Model | Implements the preference learning algorithm that converts expert rankings into a trainable reward signal. |
| RL Optimization | Proximal Policy Optimization (PPO) [59], custom RL libs | Optimizes the generative model's policy to maximize the reward from the trained reward model. |
Results and Discussion
Quantitative Performance Gains
The integration of expert feedback via RLHF and active learning paradigms yields substantial, measurable benefits in drug discovery campaigns:
- Efficiency: The hybrid 3D-QSAR/FEP active learning protocol achieved its results using less than 20% of the computational resources required for a full FEP screen, while improving the hit rate by 2.5-fold [63].
- Exploration: SynGFN demonstrated a ~70x increase in chemical space coverage compared to baseline generative models at key similarity thresholds, enabling the discovery of truly novel scaffolds [64].
- Accuracy: Active learning strategies have demonstrated up to a six-fold improvement in hit discovery over traditional, non-iterative screening methods in low-data scenarios [62]. Predictive model accuracy can also be significantly boosted; a dynamic sampling strategy optimized with active learning improved Random Forest and CNN model accuracy for toxicity prediction by 25.68% and 23.92%, respectively [66].
Addressing Bias and Scalability
A significant challenge in HITL systems is the inherent bias and inconsistency of human feedback. Research indicates that biased human feedback can reduce reinforcement learning performance by nearly 94% in terms of Average Episodic Reward [67]. The Expert-RLHF framework mitigates this through several mechanisms:
- Hybrid Bias Correction: The LLM-HFBF (Human Feedback Bias Flagging) framework uses LLMs to identify and correct for biases in human feedback before it is incorporated into reward shaping [67].
- Multi-Annotator Consensus: Aggregating feedback from multiple experts helps smooth out individual biases and inconsistencies.
- Data Efficiency: Modern RLHF research focuses on "Truncated Preference Data" and other methods to maximize the value of each human data point, mitigating the scalability issues posed by the high cost of expert time [61].
The diagram below illustrates the strategic advantage of this integrated approach over traditional methods in navigating the vast chemical space toward optimal regions.
Figure 2: Strategic Advantage of Expert-RLHF in Chemical Space Navigation
Wet-Lab Validation and Real-World Impact
The ultimate test of any computational framework is its performance in real-world experimental validation. In the case of SynGFN, generated candidates for the GluN1/GluN3A target were synthesized and tested [64]. The results were compelling:
- 10 molecules were selected for synthesis based on the model's output and proposed routes.
- 6 of the 10 compounds exhibited significant inhibitory activity.
- The most potent compound demonstrated an IC50 of 2.68 μM.
- Critically, the synthetic routes provided by SynGFN were directly executable in the lab, doubling synthetic efficiency compared to standard de novo design [64].
This successful translation from in-silico design to experimentally validated bioactive compounds underscores the practical reliability and potential of advanced HITL frameworks like Expert-RLHF to accelerate the DMTA (Design-Make-Test-Analyze) cycle in drug discovery.
The integration of expert feedback directly into reinforcement learning loops via RLHF represents a maturing paradigm for tackling complex optimization problems in scientific discovery. By combining the exploratory power of generative models and active learning with the nuanced, implicit knowledge of domain experts, the Expert-RLHF framework enables a more efficient and targeted navigation of vast design spaces, such as those encountered in drug discovery. Quantitative results across multiple studies confirm dramatic improvements in efficiency, coverage, and success rates. As methodologies for bias correction and data efficiency continue to advance [61] [67], the role of the human-in-the-loop is poised to evolve from a mere data annotator to a collaborative guide, steering computational power toward solutions that are not only high-performing but also practical and innovative. This synergy between human expertise and artificial intelligence is set to become a cornerstone of next-generation scientific research.
Benchmarking Performance: Validation, Case Studies, and Comparative Analysis
Active learning (AL) has emerged as a powerful computational strategy for efficiently navigating the vastness of chemical space in drug discovery and materials science. By iteratively selecting the most informative compounds for evaluation, AL aims to maximize model performance while minimizing resource-intensive experiments or calculations. The success of these campaigns, however, is contingent upon the rigorous application of quantitative metrics that capture not only predictive accuracy but also computational efficiency and material cost. This whitepaper provides an in-depth examination of the key performance indicators (KPIs) essential for evaluating AL campaigns in chemical exploration. Framed within a broader thesis on optimizing research in this field, we detail standardized experimental protocols, visualize core workflows, and catalog essential research tools to equip scientists with a framework for quantifying and accelerating the discovery of high-performance molecules and materials.
The chemical space of potential drug-like molecules is estimated to exceed 10^60 compounds, presenting a fundamental challenge for discovery pipelines [68] [69]. Traditional high-throughput screening, whether experimental or computational, becomes prohibitively expensive and time-consuming when applied to such immense scales. Active learning addresses this by implementing an intelligent, iterative search protocol. In an AL cycle, a machine learning model is trained on an initial set of labeled data, then used to select the most promising or informative candidates from a vast unlabeled library for evaluation by an "oracle"—such as experimental measurement or rigorous physics-based calculation [1] [70]. These new data points are incorporated into the training set, and the process repeats, allowing the model to rapidly hone in on high-performing regions of chemical space.
The critical importance of this paradigm is its dramatic efficiency gains. For instance, one study demonstrated that AL could identify high-affinity phosphodiesterase 2 (PDE2) inhibitors by explicitly evaluating only a small subset of a large chemical library [1] [71]. Another campaign for catalyst development achieved a 5-fold improvement in yield and identified the optimal system in just 86 experiments from a space of nearly five billion potential combinations, reducing the environmental footprint and cost by over 90% compared to traditional approaches [70]. To accurately gauge the success of such campaigns, a standardized set of performance metrics is indispensable, guiding model selection, protocol optimization, and ultimately, the decision to transition from in silico predictions to laboratory synthesis.
Core Performance Metrics for Active Learning Campaigns
The performance of an AL campaign must be evaluated through multiple lenses. The following tables summarize the key metrics, categorized by their primary focus.
Table 1: Predictive Performance Metrics. These metrics evaluate the accuracy and robustness of the machine learning model's predictions on the chemical property of interest (e.g., binding affinity, catalytic activity).
| Metric | Formula/Definition | Interpretation in AL Context |
|---|---|---|
| Sensitivity (Recall) | ( \frac{TP}{TP + FN} ) | Measures the model's ability to identify true active compounds (e.g., binders, potent inhibitors) from the chemical library. High sensitivity ensures valuable hits are not missed [69]. |
| Precision | ( \frac{TP}{TP + FP} ) | Measures the fraction of model-predicted actives that are true actives. High precision increases the success rate of experimental follow-up by enriching for true positives [69]. |
| F1-Score | ( 2 \times \frac{Precision \times Recall}{Precision + Recall} ) | The harmonic mean of precision and recall, providing a single metric to balance the trade-off between the two [72]. |
| Prediction Error Rate | ( \frac{FP + FN}{Total Predictions} ) | In conformal prediction frameworks, this is directly controlled by the significance level (ε), ensuring validity [69]. |
| Root Mean Square Error (RMSE) | ( \sqrt{\frac{1}{N} \sum{i=1}^{N} (yi - \hat{y}_i)^2} ) | Standard metric for the accuracy of continuous property predictions (e.g., binding free energy, reaction energy) [1]. |
Table 2: Efficiency & Cost Metrics. These metrics quantify the resource savings and operational efficiency gained by the AL approach.
| Metric | Formula/Definition | Interpretation in AL Context |
|---|---|---|
| Computational Cost Reduction | ( 1 - \frac{\text{Number of compounds evaluated with AL}}{\text{Number of compounds in full library}} ) | A direct measure of computational savings. One virtual screening study reported a reduction of over 1,000-fold by evaluating only 0.1% of a multi-billion compound library [73] [69]. |
| Experimental Cost & Footprint Reduction | Reduction in number of synthesis and testing experiments required. | In catalyst development, an AL campaign achieved >90% reduction in environmental footprint and costs by requiring only 86 experiments instead of hundreds or thousands [70]. |
| Efficiency (in Conformal Prediction) | ( \frac{\text{Number of single-label predictions}}{\text{Total number of predictions}} ) | The fraction of compounds in the library that receive a definitive (active/inactive) label from the model. A higher efficiency means a smaller, more focused library for final evaluation [69]. |
| Number of Iterations to Convergence | The number of AL cycles required to meet a pre-defined performance target (e.g., finding a compound with potency >X). | Measures the speed of the campaign. Fewer iterations indicate a more effective data selection strategy. |
Table 3: Chemical Performance Metrics. These are the ultimate experimental outcomes that the AL campaign is designed to optimize.
| Metric | Formula/Definition | Application Example |
|---|---|---|
| Binding Affinity | Free energy of binding (ΔG) or inhibitory concentration (IC50, Ki). | The primary objective in drug discovery campaigns, such as identifying PDE2 inhibitors [1] or GPCR ligands [69]. |
| Space-Time Yield (STY) | Mass of product formed per unit mass of catalyst per unit time (e.g., gHA h⁻¹ gcat⁻¹). | A key productivity metric in catalysis. An AL-optimized FeCoCuZr catalyst achieved an STY for higher alcohols of 1.1 gHA h⁻¹ gcat⁻¹, a 5-fold improvement over typical yields [70]. |
| Selectivity | Fraction of total products that is the desired product (e.g., % higher alcohol selectivity). | In multi-objective optimization, AL can minimize selectivity of undesired byproducts (e.g., CO₂, CH₄) while maintaining high productivity [70]. |
Experimental Protocols for Benchmarking AL Performance
To ensure the robustness and generalizability of an AL campaign, a standardized benchmarking protocol is essential. The following methodology outlines the key steps, drawing from proven approaches in the literature.
Protocol: Benchmarking an AL Virtual Screening Campaign
This protocol is adapted from studies that successfully applied AL to discover ligands for G protein-coupled receptors (GPCRs) and other targets [1] [69].
Data Preparation and Oracle Setup:
- Compound Library: Prepare a large, diverse chemical library (e.g., millions to billions of molecules) in a suitable molecular format. For example, the Enamine REAL space is a common source of make-on-demand compounds [69].
- Molecular Representation: Generate fixed-length vector representations for each compound. Common choices include:
- Morgan Fingerprints (ECFP4): Substructure-based circular fingerprints that are highly effective for virtual screening [69].
- Continuous Data-Driven Descriptors (CDDD): Dense latent representations from autoencoders [69].
- Graph Neural Network Embeddings (e.g., GINFP): Self-supervised embeddings that capture molecular substructure information [72].
- Define the Oracle: Establish the ground-truth method that will evaluate selected compounds. This could be:
Initial Model Training and AL Loop Configuration:
- Initial Training Set: Randomly select a small subset (e.g., 10,000-1,000,000 compounds) from the full library and obtain their scores from the oracle [69].
- Model Training: Train a machine learning classifier (e.g., CatBoost, Deep Neural Network, Gaussian Process) to predict the oracle score (or a binary active/inactive label) based on the molecular representation.
- Define Acquisition Function: This function guides the selection of compounds in each AL cycle. Common strategies include [1]:
- Greedy: Selects the top predicted binders (pure exploitation).
- Uncertainty: Selects compounds with the highest prediction uncertainty (pure exploration).
- Mixed: Selects compounds that are both high-scoring and uncertain (balancing exploitation and exploration).
Iterative Active Learning Cycle:
- Prediction: Use the trained model to predict scores/labels for the entire remaining library.
- Selection: Apply the acquisition function to choose a batch of compounds (e.g., 100-1,000) for evaluation by the oracle.
- Update: Add the new compound-oracle score pairs to the training dataset.
- Re-training: Update the machine learning model with the expanded training set.
- Repeat this cycle for a fixed number of iterations or until performance converges.
Performance Assessment:
- Throughout the cycles, track the metrics listed in Section 2 (e.g., Sensitivity, Precision, Computational Cost Reduction) against a held-out test set or the full, known library if available.
- The final model is used to select top candidates for experimental validation (e.g., synthesis and biological testing).
Workflow Visualization
The diagram below illustrates the iterative closed-loop feedback system of a standard AL campaign for chemical space exploration.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful implementation of an AL campaign relies on a suite of computational tools and chemical resources. The following table details key components.
Table 4: Essential Tools for Active Learning Campaigns in Drug Discovery.
| Category | Item/Software | Function & Application |
|---|---|---|
| Chemical Libraries | Enamine REAL, ZINC | Provide ultra-large, synthetically accessible compound collections for virtual screening, often containing billions of molecules [69]. |
| Molecular Representation | RDKit, CDDD, GINFP | Software and algorithms for generating molecular fingerprints and continuous vector representations that serve as input for machine learning models [72] [1] [69]. |
| Oracle Calculators | Schrödinger FEP+, Molecular Docking (Glide), Alchemical Free Energy Calculations | Physics-based computational methods that provide high-quality training labels (e.g., binding affinities) for the machine learning model [1] [73] [69]. |
| Machine Learning Frameworks | CatBoost, Gaussian Process, Deep Neural Networks (DNNs) | Core algorithms used to build predictive models. CatBoost, in particular, has been noted for its optimal balance of speed and accuracy in virtual screening [69]. |
| Active Learning & Optimization | Bayesian Optimization (BO), Conformal Prediction (CP) | BO guides the selection of experiments by balancing exploration and exploitation. CP provides calibrated prediction intervals, allowing control over error rates [69] [70]. |
The strategic application of active learning is reshaping the landscape of chemical space exploration by turning the computationally intractable into the efficiently manageable. The quantitative metrics and standardized protocols outlined in this whitepaper provide a foundational framework for researchers to design, execute, and critically evaluate their own AL campaigns. By moving beyond simple predictive accuracy to encompass metrics of efficiency, cost, and ultimate experimental performance, scientists can fully leverage the power of AL to accelerate the discovery of novel therapeutics, catalysts, and functional materials. As these methodologies continue to mature and integrate more deeply with experimental workflows, they promise to significantly shorten the path from conceptual design to validated breakthrough.
The exploration of vast chemical spaces is a fundamental challenge in modern drug discovery. While High-Throughput Virtual Screening (HTVS) has established itself as a traditional computational workhorse for evaluating massive compound libraries, Active Learning (AL) emerges as a strategic, data-efficient paradigm. This whitepaper provides a technical benchmark of these two approaches, demonstrating that AL frameworks achieve robust performance in hit identification and lead optimization while requiring up to 73.3% less labeled data than traditional methods [17]. By integrating machine learning with strategic sampling, AL enables targeted navigation of chemical space, offering a powerful alternative to the brute-force computational expenditure of conventional HTVS.
Drug discovery is fundamentally a search problem within an astronomically large chemical space, estimated to contain up to 10^60 drug-like compounds [1]. Computational methods are indispensable for triaging these spaces, yet they face their own scalability and efficiency challenges. For decades, High-Throughput Virtual Screening (HTVS) has been a cornerstone of computational chemistry, leveraging automation and molecular docking to rapidly score millions of compounds against a target [74] [75].
The emergence of artificial intelligence (AI) has catalyzed a paradigm shift. Active Learning (AL), a subfield of AI, introduces an iterative, closed-loop workflow where machine learning models selectively query the most informative data points for labeling, dramatically reducing the number of computationally expensive simulations or experiments required [17] [1]. This paper benchmarks AL against traditional HTVS within the context of chemical space exploration, evaluating their respective methodologies, performance, and applicability to modern drug discovery challenges.
Methodological Comparison
The core distinction between HTVS and AL lies in their approach to sampling and evaluation. The following workflows illustrate their fundamental processes.
High-Throughput Virtual Screening (HTVS) Workflow
HTVS operates as a linear, exhaustive screening pipeline. It prioritizes breadth, systematically evaluating every compound in a library through computational simulations.
Active Learning (AL) for Chemical Space Workflow
AL employs an iterative, intelligent cycle. The model learns from a small initial dataset and progressively refines its search by querying an "oracle" for the most uncertain or promising candidates.
Performance Benchmarking and Quantitative Comparison
Retrospective and prospective studies provide quantitative evidence of the comparative performance of AL and HTVS strategies.
Key Performance Metrics from Recent Studies
Table 1: Benchmarking performance of Active Learning and HTVS approaches.
| Study Focus | Method | Key Performance Metrics | Data Efficiency |
|---|---|---|---|
| Toxicity Prediction (TDCs) [17] | Active Stacking-DL with Strategic Sampling | MCC: 0.51, AUROC: 0.824, AUPRC: 0.851 | Achieved high performance with ~73% less labeled data than full-data models. |
| PDE2 Inhibitor Identification [1] | AL with Alchemical Free Energy Oracle | Efficiently identified high-affinity binders; performance dependent on selection strategy. | Required explicit evaluation of only a small subset of a large chemical library. |
| Traditional Virtual Screening [74] [75] | HTVS (Molecular Docking) | High dependency on scoring function accuracy; prone to false positives without secondary screens. | Requires full-library evaluation, leading to high computational cost per cycle. |
Analysis of Compound Selection Strategies in AL
The efficacy of an AL pipeline is highly dependent on the query strategy used to select compounds. Research on PDE2 inhibitors benchmarked several approaches [1]:
- Greedy Selection: Chooses only the top predicted binders each round. It rapidly improves average affinity but risks getting trapped in local minima.
- Uncertainty Selection: Selects ligands with the largest prediction uncertainty. Excellent for exploration and model improvement.
- Mixed Strategy: First identifies the top 300 predicted binders, then selects the 100 with the most uncertain predictions from this pool. This hybrid approach effectively balances exploration and exploitation, making it one of the most robust strategies [1].
Experimental Protocols and Implementation
Protocol for an Active Learning Cycle
Implementing a robust AL cycle for chemical space exploration involves several critical steps, as demonstrated in prospective studies [1]:
Initialization & Data Preparation:
- Generate a large, diverse in silico compound library.
- Select an initial small training set via weighted random sampling to ensure diversity. Probability of selection can be inversely proportional to the number of structurally similar molecules already in the set.
Molecular Representation (Feature Engineering):
- Encode compounds into fixed-size vectors for ML. Common representations include:
- 2D/3D Descriptors: Constitutional, electrotopological, and molecular surface area descriptors calculated with tools like RDKit [1].
- Interaction-Based Features: PLEC fingerprints or residue-ligand interaction energies from molecular dynamics [1].
- MedusaNet-inspired Voxels: Grid-based representation of ligand atoms within the binding site [1].
- Encode compounds into fixed-size vectors for ML. Common representations include:
Model Training & The Oracle:
- Train an ML model (e.g., regression model, graph neural network) on the current labeled training set to predict bioactivity (e.g., binding affinity).
- The oracle is the method used to obtain the "true" label for selected compounds. High-accuracy but computationally expensive alchemical free energy calculations are an ideal oracle for optimizing binding affinity [1].
Iterative Active Learning Loop:
- Use the trained model to predict on the entire unlabeled pool.
- Apply a selection strategy (e.g., mixed strategy) to choose the most informative batch of compounds.
- Query the oracle for these compounds' labels.
- Add the newly labeled compounds to the training set and repeat from Step 3 until a stopping criterion is met (e.g., budget exhausted or desired affinity is found).
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key software and computational tools for implementing AL and HTVS workflows.
| Tool / Resource | Type | Primary Function in Workflow |
|---|---|---|
| ZINC/ChEMBL [74] | Database | Provides access to millions of commercially available compounds with annotated data for virtual screening. |
| RDKit [1] | Cheminformatics | Open-source toolkit for calculating molecular descriptors, fingerprints, and generating 3D conformations. |
| AutoDock/Glide [74] | Docking Software | Core engines for HTVS, performing molecular docking and scoring for large compound libraries. |
| GROMACS [74] [1] | Molecular Dynamics | Software for running MD simulations to refine binding poses and calculate interaction energies for feature generation. |
| pmx [1] | Free Energy Toolbox | Used for preparing hybrid topologies and running alchemical free energy calculations, serving as a high-accuracy oracle. |
| DeepChem [74] | ML Library | Deep learning toolkit specifically designed for chemical data, useful for building AL prediction models. |
Benchmarking analyses firmly position Active Learning not as a mere replacement for HTVS, but as a sophisticated, complementary paradigm. AL addresses critical inefficiencies of traditional virtual screening by dramatically reducing computational costs and enabling a more intelligent, adaptive exploration of chemical space.
The future of chemical space exploration lies in hybrid frameworks that leverage the strengths of both approaches. Promising directions include the integration of multimodal data (genomic, proteomic) [74], the use of federated learning to collaborate on data without sharing it [74], and the advancement of explainable AI (XAI) to build trust and provide mechanistic insights into model predictions [74]. As these technologies mature, the iterative, data-efficient principles of AL are poised to become central to the next generation of drug discovery.
Prospective validation represents the definitive step in computational drug discovery, where computationally identified "hits" are experimentally tested to confirm biological activity. This process moves beyond retrospective benchmarks to provide real-world evidence of a screening method's efficacy, bridging the gap between in silico predictions and tangible therapeutic candidates. Within the broader context of active learning chemical space exploration, prospective validation serves as the essential feedback mechanism that closes the loop between prediction and experimental reality. As drug discovery faces increasing pressure to improve efficiency and reduce costs, the integration of active learning with prospective experimental validation has emerged as a transformative paradigm, enabling researchers to navigate massive chemical spaces with unprecedented efficiency while generating experimentally confirmed hits for further development [76] [77].
The fundamental challenge in hit identification lies in the astronomical size of drug-like chemical space, estimated to contain 10^60 possible compounds, making exhaustive exploration impossible [76]. Traditional virtual screening methods have made significant inroads, but often rely on static models and predetermined compound libraries. In contrast, active learning systems incorporate experimental feedback directly into the exploration process, creating a continuous cycle of prediction, testing, and model refinement that progressively focuses on the most promising regions of chemical space. This article provides a comprehensive technical examination of prospective validation methodologies, experimental protocols, and the research infrastructure required to successfully confirm computed hits within modern active learning frameworks.
Core Principles and Definitions
Key Concepts in Hit Identification
Prospective validation refers to the process of experimentally testing compounds selected by computational methods without prior knowledge of their activity, providing the most credible assessment of a screening method's real-world performance [77]. This contrasts with retrospective validation, where methods are tested on known actives and decoys. Hit identification encompasses the entire process of discovering initial active compounds against a biological target, which then serve as starting points for medicinal chemistry optimization [78]. The criteria for designating a compound as a "hit" vary significantly across studies but typically involve specific thresholds of biological activity such as IC50, Ki, or percentage inhibition [78].
Ligand efficiency (LE) has emerged as a critical metric in hit assessment, normalizing biological activity to molecular size to enable more meaningful comparison between compounds [78]. This is particularly important in fragment-based screening approaches. Chemical space exploration refers to the systematic investigation of possible molecular structures and properties to identify regions containing compounds with desired biological activities. When combined with active learning, this becomes an iterative process where experimental results inform subsequent computational exploration, creating a continuous cycle of improvement [76].
The Active Learning Cycle in Chemical Space Exploration
Active learning represents a fundamental shift from traditional screening approaches by incorporating experimental feedback directly into the chemical exploration process. As illustrated in the workflow diagram, this creates a continuous cycle where each iteration refines the model's understanding of the structure-activity relationship. The system begins with an initial, often small, set of experimental data which seeds the machine learning model. The model then prioritizes compounds from the vast chemical space for experimental testing, with newly generated data feeding back to improve the model's predictive capabilities for subsequent cycles [76].
This approach dramatically reduces the experimental burden required to identify hits. One recent demonstration successfully explored a virtual search space of one million potential battery electrolytes starting from just 58 initial data points [76]. While this example comes from materials science, the same principles apply directly to drug discovery, where the efficiency gains can substantially accelerate early-stage discovery campaigns.
Methodologies for Prospective Validation
Integrated Computational-Experimental Workflows
Successful prospective validation requires tight integration between computational prediction and experimental confirmation. A proven workflow begins with target evaluation and selection using tools like SpectraView, which employs knowledge graphs to assess biological targets based on scientific and commercial considerations [77]. This is followed by virtual screening using methods such as HydraScreen, a deep learning-based scoring function that predicts protein-ligand affinity and pose confidence [77]. The top-ranked compounds then proceed to experimental testing in automated robotic cloud labs like Strateos, which enable highly reproducible data generation with precise control of experimental conditions [77].
This integrated approach was prospectively validated in a study targeting IRAK1, where the deep learning model identified 23.8% of all hits within the top 1% of ranked compounds—significantly outperforming traditional virtual screening methods [77]. The workflow's effectiveness stems from its closed-loop design, where experimental results can potentially feed back into model refinement, though this active learning component was not explicitly highlighted in the IRAK1 case study.
Experimental Design and Hit Confirmation Protocols
Robust experimental design is crucial for reliable prospective validation. Primary screening typically employs concentration-response endpoints (IC50, EC50, Ki, or Kd) or single-concentration percentage inhibition to determine initial activity [78]. For the IRAK1 validation study, researchers used a diverse library of 46,743 commercially available compounds characterized by scaffold diversity and favorable physicochemical properties, with interference compounds systematically removed [77].
Hit confirmation requires orthogonal assays to validate initial findings. As analyzed across multiple virtual screening studies, this typically includes secondary assays to confirm activity (74% of studies), binding assays to demonstrate direct target engagement (74%), and counter screens to assess selectivity (116%) [78]. These layered confirmation steps ensure that identified hits represent genuine biological activity rather than assay artifacts or non-specific effects.
Table 1: Hit Identification Criteria in Virtual Screening Studies (2007-2011)
| Hit Calling Metric | Studies Using Metric | Typical Activity Range | Ligand Efficiency Application |
|---|---|---|---|
| IC50 | 30 studies | 1-25 μM | Rarely used |
| % Inhibition | 85 studies | Varies by study | Not applied |
| Ki/Kd | 4 studies | Sub-μM to low μM | Occasionally considered |
| EC50 | 4 studies | Low μM | Not typically used |
Source: Adapted from analysis of 421 virtual screening studies [78]
Quantitative Assessment of Virtual Screening Performance
Hit Rates and Ligand Efficiency Analysis
Comprehensive analysis of virtual screening studies reveals important patterns in hit identification performance. Across 421 studies published between 2007-2011, only approximately 30% reported a clear, predefined hit cutoff, with significant variation in activity thresholds used to designate compounds as hits [78]. The majority of studies (136) used activity cutoffs in the 1-25 μM range, while surprisingly, 56 studies used 100-500 μM and 25 studies used >500 μM as their initial activity cutoff [78].
Hit rates varied considerably based on screening library size and methodology. Studies screening libraries of 100,001-1,000,000 compounds typically tested 10-50 compounds experimentally, while those with smaller libraries (<1,000 compounds) tested 1-10 compounds on average [78]. The application of ligand efficiency metrics remains underutilized in hit identification, with only fragment-based screens consistently employing this size-targeted efficiency measure [78].
Table 2: Performance Metrics in Prospective Virtual Screening Validation
| Screening Method | Hit Rate Performance | Key Advantages | Limitations |
|---|---|---|---|
| Traditional Virtual Screening | Variable; highly dependent on target and library | Well-established methodology | Limited learning from experimental data |
| Deep Learning (HydraScreen) | 23.8% of hits in top 1% of ranked compounds [77] | High early enrichment; pose confidence scoring | Requires substantial training data |
| Active Learning with Minimal Data | Identified 4 novel electrolytes from 58 starting points [76] | Extreme efficiency with limited data | Potential for model bias in early cycles |
| Structure-Based Docking | Widely used but variable performance | Handles novel chemotypes; structural insights | Scoring function inaccuracies |
Economic Considerations in Hit Confirmation
The decision of how many hits to send for confirmatory testing represents an economic optimization problem rather than purely a statistical one [79]. Traditional statistical approaches aim to control false discovery rates at predetermined tolerances but neglect local economic context, potentially leading to suboptimal experimental strategies [79]. Economic frameworks that meaningfully quantify the trade-off between true and false positives can identify economically optimal experimental strategies, with retrospective simulations demonstrating the identification of hundreds of additional actives that had been erroneously labeled inactive in real-world screening experiments [79].
This economic perspective is particularly relevant in active learning approaches, where the cost of experimental testing directly influences which compounds are selected for each iteration. By explicitly incorporating testing costs and expected value of information, active learning systems can maximize the efficiency of the entire discovery process rather than simply maximizing the number of hits identified.
Research Reagent Solutions and Experimental Infrastructure
Essential Research Tools for Prospective Validation
Table 3: Key Research Reagent Solutions for Hit Identification and Validation
| Tool/Technology | Function | Application in Prospective Studies |
|---|---|---|
| SpectraView | Target evaluation using knowledge graphs | Data-driven assessment of biological targets based on scientific and commercial criteria [77] |
| HydraScreen | Deep learning-based virtual screening | Predicts protein-ligand affinity and pose confidence; outperformed traditional methods in IRAK1 study [77] |
| Strateos Cloud Lab | Automated robotic experimentation | Enables highly reproducible HTS with precise environmental control and remote monitoring [77] |
| 47k Diversity Library | Curated compound collection | 46,743 commercially available compounds with scaffold diversity and favorable physicochemical properties [77] |
| Knowledge Graph Systems | Biomedical data integration | Comprehensive data resource with 12 entity types for contextual target and compound analysis [77] |
Automated Experimental Platforms
The integration of automated robotic cloud labs has transformed prospective validation by enabling highly reproducible, remotely operated experimental workflows. Platforms like the Strateos Cloud Lab consist of automated chemistry and biology workstations integrated with inventory management, data generation, and data management systems [77]. Experiments are coded in Autoprotocol, an open-source standard that coordinates instrument actions based on scientific intent, allowing researchers to configure experiments remotely, monitor automated execution, and access real-time experimental data in a closed-loop fashion [77].
This automation is particularly valuable in active learning cycles, where rapid experimental turnaround is essential for maintaining momentum in the iterative exploration process. The ability to quickly test computational predictions and feed results back into the model enables more cycles of learning within practical timeframes, significantly accelerating the overall discovery process.
Visualization of Workflows and Signaling Pathways
Active Learning Cycle for Hit Identification
Active Learning Cycle for Hit Identification
Integrated Computational-Experimental Workflow
Integrated Computational-Experimental Workflow
Case Study: Prospective Validation of IRAK1 Inhibitors
Experimental Protocol and Methodologies
A recent prospective validation study targeting IRAK1 provides a compelling case study in integrated hit identification [77]. The research employed a comprehensive workflow beginning with target evaluation using SpectraView, which analyzed IRAK1 within a knowledge graph containing 12 entity types (Disease, Target, Mechanism, Compound, etc.) drawn from over 34 million PubMed abstracts and 90 million patents [77]. Virtual screening was performed using HydraScreen, a convolutional neural network-based deep learning framework trained on more than 19,000 protein-ligand pairs and 290,000 docked conformations [77].
Experimental validation utilized the Strateos robotic cloud lab, with compounds stored as 10 mM stocks in DMSO and 10 nL transferred to screening plates using a Beckman Echo system [77]. The study employed a diverse library of 46,743 compounds characterized by scaffold diversity and favorable physicochemical attributes, with Pan Assay Interference Compounds (PAINS) systematically removed [77]. For compounds with undefined stereocenters, researchers generated all possible stereoisomers (maximum 16) and computed final scores by averaging across stereoisomers to mimic racemic-averaged experimental results [77].
Results and Performance Metrics
The prospective validation demonstrated exceptional performance, with the deep learning model identifying 23.8% of all IRAK1 hits within the top 1% of ranked compounds [77]. The study identified three potent (nanomolar) scaffolds from the compound library, two of which represented novel candidates for IRAK1 with promise for future development [77]. This performance significantly exceeded traditional virtual screening techniques while providing additional capabilities such as ligand pose confidence scoring [77].
Notably, the integration of automated experimentation enabled highly reproducible data generation with precise control of experimental conditions, essential for reliable hit confirmation [77]. The entire process—from target evaluation through experimental confirmation—showcased how AI-driven virtual screening combined with automated laboratories can accelerate early-stage drug discovery while reducing experimental costs [77].
Future Directions and Implementation Recommendations
The integration of active learning with prospective validation represents the frontier of efficient chemical space exploration. Future developments will likely focus on fully generative AI models that create novel molecular structures beyond existing chemical databases, potentially discovering scaffolds previously unimagined by medicinal chemists [76]. Additionally, multi-objective optimization approaches that simultaneously evaluate multiple criteria—including potency, selectivity, safety, and developability properties—will be essential for identifying compounds with true translational potential [76].
For research teams implementing these approaches, key recommendations emerge from successful case studies. First, establish a closed-loop infrastructure connecting computational prediction with automated experimental validation. Second, implement economic frameworks to optimize the trade-off between experimental costs and information gain. Third, apply size-targeted ligand efficiency metrics early in hit identification to select compounds with optimal optimization potential [78]. Finally, embrace iterative active learning cycles that progressively focus exploration on the most promising regions of chemical space, dramatically increasing the efficiency of the hit discovery process.
As these methodologies mature, the integration of prospective validation within active learning frameworks will continue to transform early drug discovery, enabling more efficient navigation of massive chemical spaces while generating experimentally confirmed hits with enhanced prospects for successful development into therapeutic candidates.
The escalating complexity of drug discovery, particularly in targeting protein-protein interactions (PPIs) and kinase enzymes, has intensified the need for more efficient research methodologies. Efficiency in this context transcends mere speed, encompassing the maximal biochemical effect achieved from minimal molecular interventions—whether measured as binding energy per unit of interface area in PPIs or degradation efficacy per small molecule in kinase inhibition. This paradigm aligns with the emerging framework of active learning chemical space exploration, where artificial intelligence (AI) guides experimentation to navigate vast molecular possibilities with minimal data input [76]. This case study analyzes quantitative efficiency gains in these domains, framing them within a broader thesis on optimized research strategies that are revolutionizing therapeutic development.
Efficiency in Protein-Protein Interaction Interfaces
Quantitative Framework for PPI Binding Efficiency
Protein-protein interactions are fundamental to cellular processes, and their modulation offers significant therapeutic potential. A critical metric for evaluating these interactions is binding efficiency, defined as the binding energy generated per square Ångstrom (Ų) of surface area buried at the contact interface [80]. This concept extends the established principle of "ligand efficiency" from small-molecule drug discovery to the more complex realm of PPIs. Analysis of a diverse set of 144 protein-protein complexes has revealed that the most efficient PPI complexes can generate approximately 20 cal·mol⁻¹/Ų of binding energy [80]. Furthermore, studies indicate a minimum contact area of approximately 500 Ų is necessary to form a stable complex, as this provides sufficient interaction energy to overcome the entropic cost of co-localizing two proteins from a 1 M solution [80].
Case Study: Binding Efficiency Across TNF/TNFR Family Complexes
The tumor necrosis factor (TNF) superfamily and their receptors provide an illustrative model for studying binding efficiency due to their conserved binding mode across interfaces of varying sizes. Research has quantified single-site binding affinities and calculated binding efficiencies for several family members, revealing striking variations.
Table 1: Binding Affinities and Efficiencies in Select TNF/TNFR Complexes [80]
| Ligand-Receptor Pair | Binding Affinity (K_D) | Interface Properties | Binding Efficiency (cal·mol⁻¹/Ų) | Relative Efficiency (%) |
|---|---|---|---|---|
| TNFα/TNFR1 | 1.4 ± 0.4 nM | Large interface | Data not specified | 44-49% of maximum |
| BAFF/BR3 | High affinity | Compact, single CRD fragment | ~16 (est. from 80% of max) | ~80% of maximum |
| TWEAK/Fn14 | 70 ± 10 nM | Small interface | Data not specified | High |
A key finding is that smaller receptors, such as BR3 (containing only a fragment of a cysteine-rich domain), can achieve remarkably high binding efficiencies—up to 80% of the proposed maximum achievable value [80]. In contrast, larger receptors like TNFR1, despite their more extensive contact surfaces, operate at only 44-49% of this efficiency limit [80]. This demonstrates that interface size does not directly correlate with efficiency; rather, the spatial arrangement and chemical complementarity of interfacial residues are paramount.
Experimental Protocol for Determining Binding Efficiency
Objective: To quantify the binding affinity and calculate the binding efficiency of a protein-protein interaction.
Materials:
- Purified recombinant proteins (e.g., TNFα, TNFR1) [80]
- Surface Plasmon Resonance (SPR) instrument (e.g., Biacore)
- CMS sensor chips for immobilization
- HBS-EP running buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20, pH 7.4)
Methodology:
- Ligand Immobilization: One binding partner (the ligand, e.g., TNFα) is covalently immobilized on the surface of a CMS sensor chip using standard amine coupling chemistry.
- Analyte Binding: The second partner (the analyte, e.g., TNFR1) is injected over the chip surface at a series of known concentrations in the running buffer.
- Data Collection: The SPR instrument measures the association and dissociation of the analyte to the immobilized ligand in real time, generating sensorgrams.
- Affinity Calculation: The resulting sensorgrams are fitted to a 1:1 Langmuir binding model using the instrument's software. The equilibrium dissociation constant (KD) is derived from the ratio of the dissociation rate constant (kd) to the association rate constant (ka). The standard free energy of binding (ΔG°) is then calculated using the equation ΔG° = RT ln(KD), where R is the gas constant and T is the temperature in Kelvin [80].
- Interface Area Determination: The solvent-accessible surface area (ASA) buried upon complex formation (ΔASA) is determined from a co-crystal structure of the complex using computational tools like NACCESS.
- Efficiency Calculation: The binding efficiency is finally calculated as |ΔG°| / ΔASA, reported in units of cal·mol⁻¹/Ų [80].
AI-Driven Exploration of Chemical Space for Efficiency Gains
The Active Learning Paradigm in Chemical Discovery
A transformative approach to achieving efficiency in early-stage discovery is AI-driven active learning. This strategy enables the exploration of massive chemical spaces with minimal experimental data, dramatically accelerating the identification of promising candidates. A pioneering study successfully identified four high-performing battery electrolytes from a virtual search space of one million potential molecules by starting with just 58 initial data points [76]. The core of this methodology is an iterative loop where the AI model suggests experiments, the results of which are fed back to refine its predictions.
Experimental Protocol for Active Learning in Molecular Screening
Objective: To efficiently identify molecules with desired properties from a vast chemical space using an AI-guided active learning loop.
Materials:
- A defined virtual chemical library (e.g., 1 million electrolyte candidates) [76]
- An initial, small set of experimental data ("seed data") for model training
- High-throughput or automated experimental setup for property validation (e.g., battery cycling for cycle life) [76]
- Computational resources for AI model training and inference
Methodology:
- Model Initialization: Train an initial machine learning model on the small seed dataset to predict molecular properties.
- Candidate Selection: The model screens the vast virtual library, predicting performance and associated uncertainty for each candidate. It then prioritizes a batch of candidates that are either predicted to be high-performing or reside in regions of high uncertainty (exploration vs. exploitation).
- Experimental Validation: The selected candidates are synthesized and tested experimentally (e.g., by building and cycling batteries) to obtain ground-truth performance data [76].
- Model Retraining: The new experimental data is added to the training set, and the model is retrained, improving its accuracy for the next iteration.
- Loop Iteration: Steps 2-4 are repeated for multiple cycles (e.g., 7 campaigns of ~10 experiments each) until candidates meeting the target criteria are identified [76].
AI Active Learning Loop
Efficiency Through Targeted Protein Degradation in Kinase Inhibition
Beyond Inhibition: Induced Degradation as an Efficient Strategy
A profound efficiency gain in kinase pharmacology is the shift from mere inhibition to targeted degradation. Conventional kinase inhibitors block ATP-binding activity but leave the protein scaffold intact, often leading to compensatory resistance mechanisms. A more efficient approach is to use small molecules to induce the complete removal of the kinase protein from the cell. A systematic study profiling 1,570 kinase inhibitors against 98 kinases revealed that 232 compounds caused destabilization of at least one kinase, with 160 unique selective kinase-compound pairs identified as degraders [81]. This phenomenon, termed "supercharging," occurs when inhibitors induce a kinase conformation that is more efficiently recognized and cleared by the cell's native proteolytic systems [81].
Mechanisms of Inhibitor-Induced Kinase Degradation
The study elucidated several mechanistic principles through which inhibitors supercharge endogenous degradation circuits:
- Modulation of Kinase Activity: Inhibitors can lock kinases into conformations that expose degrons (peptide sequences recognized by ubiquitin ligases). For example, certain inhibitors induce the degradation of LYN kinase this way [81].
- Perturbation of Intracellular Localization: Redirecting a kinase away from its stabilizing microenvironment can promote degradation, as observed for BLK kinase [81].
- Induction of Higher-Order Assemblies: Some inhibitors trigger the formation of kinase aggregates or complexes that are targeted for autophagic or proteasomal clearance, exemplified by RIPK2 [81].
- Chaperone Deprivation: Many kinases, particularly mutants, are clients of the HSP90 chaperone complex. Inhibitors like neratinib can disrupt the HER2-HSP90 interaction, leading to HER2 destabilization and degradation [81].
Table 2: Case Studies in Kinase Degradation Mechanisms [81]
| Kinase Target | Representative Inhibitor/Degrader | Primary Mechanism of Destabilization | Clinical/Experimental Context |
|---|---|---|---|
| HER2 | Neratinib, Afatinib | Chaperone Deprivation (HSP90 disruption) | Cancer |
| LYN | Specific inhibitors from screen | Inducing a degradation-prone conformational state (Activity Modulation) | Cancer, Immune signaling |
| BLK | Specific inhibitors from screen | Altering subcellular localization | B-cell signaling |
| RIPK2 | Specific inhibitors from screen | Inducing higher-order assemblies / aggregation | Inflammatory signaling |
| Mutant PI3Kα | Inavolisib, Taselisib | Mechanism beyond chaperone deprivation, involves network effects | Cancer |
Experimental Protocol for Systematic Degrader Identification
Objective: To systematically identify kinase inhibitors that induce protein degradation.
Materials:
- A panel of cell lines (e.g., K562) expressing kinases of interest as Nanoluciferase (Nluc) fusion proteins [81]
- A library of kinase inhibitors (e.g., 1,570 compounds)
- Control cell lines expressing stable (GFP-Nluc) and unstable (dGFP-Nluc) reporters
- Luminescence plate reader for quantitative abundance measurement
- Facilities for cell culture and compound handling
Methodology:
- Reporter System: Generate a stable cell panel where 98 kinase open reading frames (88 canonical, 10 mutants) are expressed as Nluc fusion proteins to report on abundance [81].
- Dynamic Compound Profiling: Treat each kinase-reporter cell line with the inhibitor library. Measure luminescence (reporting on kinase abundance) at multiple time points (e.g., 2, 6, 10, 14, 18 h) [81].
- Data Normalization and Hit Scoring: Normalize luminescence data against vehicle controls and non-kinase control cell lines to filter out global effects on transcription/translation or compound cytotoxicity. Implement a multi-tiered scoring scheme to identify compounds that cause significant downregulation of kinase levels.
- Validation and Mechanistic Studies: Confirm hits using orthogonal methods (e.g., western blotting). Investigate mechanism through co-immunoprecipitation (e.g., for HSP90 interaction), cellular imaging (for localization), or analysis of higher-order complexes.
Integrated Workflow and Research Toolkit
The convergence of PPI analysis, kinase degradation, and active learning represents a powerful, integrated workflow for modern drug discovery.
Integrated Drug Discovery Workflow
Table 3: The Scientist's Toolkit for Efficiency-Focused Research
| Research Reagent / Tool | Function and Utility | Field of Application |
|---|---|---|
| Surface Plasmon Resonance (SPR) | Quantifies binding kinetics (KD, ka, k_d) and affinity for biomolecular interactions. | PPI Binding Efficiency [80] |
| Nanoluciferase (Nluc) Fusion Reporter | Sensitive luminescent reporter for tracking dynamic changes in protein abundance in live cells. | Kinase Degradation Screening [81] |
| Active Learning AI Model | Guides iterative experimentation to explore vast chemical spaces with minimal initial data. | Chemical Space Exploration [76] |
| Co-crystal Structure Analysis | Provides atomic-resolution details of the binding interface, enabling ΔASA calculation. | PPI Interface Analysis [80] |
| Hyperbolic Graph Neural Network (e.g., HI-PPI) | Captures hierarchical information in PPI networks for improved interaction prediction. | Computational PPI Prediction [82] |
| Proteolysis-Targeting Chimeras (PROTACs) | Bifunctional molecules that recruit E3 ligases to target proteins to induce degradation. | Targeted Protein Degradation |
This analysis demonstrates that the pursuit of efficiency—whether in the form of maximizing binding energy per interface area, minimizing data requirements for chemical exploration, or achieving complete protein removal versus inhibition—is a unifying driver of innovation in biomedical research. The quantitative frameworks for PPI efficiency, the AI-driven paradigms for navigating chemical space, and the mechanistic insights into kinase degradation collectively represent a powerful toolkit for the next generation of drug discovery. Future efforts will likely focus on integrating these approaches more seamlessly, using multi-objective AI models to simultaneously optimize for binding, degradation, and drug-like properties, thereby accelerating the development of more effective and precise therapeutics.
Active learning (AL) for chemical space exploration represents a paradigm shift in computational drug discovery, enabling the efficient navigation of vast molecular landscapes estimated to contain up to 10^60 drug-like compounds [1] [13]. This iterative feedback process efficiently identifies valuable data within this immense space, making it particularly valuable given the limitations of labeled experimental data [83]. However, as research institutions and pharmaceutical companies increasingly adopt AL methodologies to accelerate development cycles, significant bottlenecks and domain boundaries have emerged that limit their full potential. This technical analysis examines the current constraints of AL in chemical exploration, synthesizing quantitative performance data, detailing experimental protocols, and identifying critical research gaps that must be addressed to advance the field.
Quantitative Analysis of Current Limitations
The implementation of active learning in chemical space exploration faces several quantifiable constraints that impact efficiency and applicability. The table below summarizes key bottleneck metrics identified from recent literature.
Table 1: Quantitative Bottlenecks in Active Learning for Chemical Space Exploration
| Constraint Category | Specific Limitation | Quantitative Impact | Domain Affected |
|---|---|---|---|
| Computational Cost | Alchemical free energy calculations as oracle | Hundreds to thousands of ligands evaluated in days [1] | Lead optimization |
| Data Efficiency | Labeled data requirements for molecular property prediction | Sparse, scarce, and expensive to generate [13] | Generalizability |
| Chemical Space Coverage | Exploration of synthetically accessible organic molecules | 6B molecules in Enamine REALSpace Dataset [13] | Model transferability |
| Model Generalization | Performance on novel molecular scaffolds | Poor generalization to molecules substantially different from training data [13] | Cross-domain applications |
Methodological Constraints in Experimental Protocols
Active Learning Cycle Implementation
The standard AL protocol for chemical space exploration follows an iterative workflow with distinct methodological constraints:
Initialization Phase (Iteration 0):
- Weighted Random Selection: Compounds are selected with probability inversely proportional to similar compounds in the dataset [1]
- Similarity Metric: Ligands are considered similar if they fall within the same bin of a 2D histogram after t-SNE embedding [1]
- Representation Dependency: Embedding constructed from 2D features (constitutional and graph descriptors with MACCS and BCUT2D fingerprints) [1]
Iterative Active Learning Phase:
- Batch Selection: Typically 100 ligands selected per iteration [1]
- Oracle Evaluation: Alchemical free energy calculations provide binding affinity data [1]
- Model Retraining: Newly acquired data incorporated into updated ML models [1]
Ligand Selection Strategies: Multiple strategies have been developed with inherent trade-offs:
- Greedy Selection: Chooses only top predicted binders each iteration, potentially limiting diversity [1]
- Uncertainty Sampling: Selects ligands with largest prediction uncertainty, promoting exploration [1]
- Mixed Strategy: Identifies 300 strongest predicted binders, then selects 100 with most uncertain predictions, balancing exploitation and exploration [1]
- Narrowing Strategy: Broad selection in first 3 iterations followed by greedy approach, using multiple models with different descriptor sets [1]
Molecular Representation Limitations
The encoding of molecular structure significantly impacts AL performance, with current methods presenting distinct constraints:
Table 2: Molecular Representation Schemes and Their Limitations
| Representation Scheme | Technical Description | Domain Constraints |
|---|---|---|
| 2D_3D Features | Combines constitutional, electrotopological, molecular surface area descriptors, and multiple molecular fingerprints [1] | Limited by descriptor completeness and computational cost |
| Atom-hot Encoding | Binding site split into 2Å voxels counting ligand atoms per element [1] | Fixed grid resolution may miss subtle interactions |
| PLEC Fingerprints | Contacts between ligand and each protein residue [1] | Protein flexibility not captured |
| MDenerg Representations | Electrostatic and van der Waals interaction energies per residue [1] | Force field accuracy dependency |
Domain Boundaries and Transferability Constraints
Chemical Space Coverage Limitations
Current AL implementations face significant boundaries in chemical domain transferability:
Training Data Homogeneity: Despite chemical space heterogeneity, existing foundation models like MIST are primarily trained on synthetically accessible organic molecules from the Enamine REALSpace dataset [13]. This creates inherent biases when applying models to:
- Organometallic compounds and inorganic complexes [13]
- Chiral molecules and stereochemical nuances [13]
- Radioactive isotopes and exotic elements [13]
Nuclear and Electronic Representation: The Smirk tokenization algorithm attempts to capture nuclear, electronic, and geometric features [13], but performance degrades when encountering molecular features underrepresented in training data.
Foundation Model Scaling Limitations
The development of scientific foundation models (SciFMs) for chemical space presents computational boundaries:
Model Scaling Laws: Current neural scaling laws assume optimal hyperparameter tuning but lack formulations for data quality impacts [13]. The MIST model family demonstrates scaling up to 1.8 billion parameters trained on 2 billion molecules [13], yet the relationship between model size, data quality, and chemical accuracy remains poorly quantified.
Compute-Optimal Training: Hyperparameter-penalized Bayesian neural scaling laws have reduced development costs by over an order of magnitude (saving ~10 petaflop-days) [13], but optimal scaling exponents for chemical domain coverage remain undefined.
Visualization of Workflows and Limitations
Active Learning Cycle with Bottlenecks
Active Learning Cycle with Computational Bottlenecks
Chemical Space Coverage Limitations
Chemical Space Coverage and Exploration Boundaries
Essential Research Reagents and Computational Tools
Table 3: Key Research Reagent Solutions for Active Learning Implementation
| Tool/Category | Specific Implementation | Function in Workflow |
|---|---|---|
| Cheminformatics | RDKit [1] | Molecular fingerprint generation, descriptor calculation, and constrained embedding |
| Free Energy Calculations | pmx [1], Gromacs [1] | Alchemical binding free energy predictions as oracle |
| Molecular Dynamics | Amber99SB*-ILDN force field [1], GAFF 1.9 [1] | Binding pose refinement and interaction energy calculation |
| Machine Learning | Scikit-learn, Custom neural networks [1] | Model training with various molecular representations |
| Molecular Representation | PLEC fingerprints [1], MedusaNet-inspired encodings [1] | Protein-ligand interaction featurization |
| Foundation Models | MIST models [13] | Transfer learning for molecular property prediction |
Active learning for chemical space exploration faces significant bottlenecks in computational cost, data efficiency, and model generalizability, with clear domain boundaries at the frontiers of chemical diversity. Current methodologies successfully navigate synthetically accessible organic compounds but struggle with transferability to underrepresented chemical domains like organometallics and complex mixtures. The integration of foundation models presents promising pathways to overcome these limitations through improved representation learning and transferable feature extraction. Future research must address the quantification of data quality in scaling laws, development of more comprehensive molecular representations, and creation of standardized benchmarking across diverse chemical domains to systematically overcome these constraints.
Conclusion
Active learning has firmly established itself as a transformative paradigm for chemical space exploration, demonstrably accelerating the discovery of novel bioactive molecules and materials. The synthesis of strategies covered—from foundational iterative loops to advanced multi-level Bayesian optimization and human-in-the-loop refinement—provides a robust framework for tackling the immense scale of molecular discovery. Key takeaways include the critical importance of strategic exploration-exploitation balance, the power of integrating physics-based oracles like free energy calculations, and the proven ability of AL to achieve up to a 1000-fold increase in screening efficiency. Looking forward, the integration of AL with automated synthesis and testing platforms promises a future of closed-loop, autonomous discovery systems. The ongoing development of more accurate property predictors and generalizable models will further extend the reach of AL, enabling the routine discovery of structurally novel, synthetically accessible, and therapeutically 'beautiful' molecules that push the boundaries of biomedical research.
References
- 1. Chemical Space Exploration with Active Learning and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9558370/]
- 2. Navigating large chemical spaces in early-phase drug ... [https://www.sciencedirect.com/science/article/pii/S0959440X23000520]
- 3. Chemical space visual navigation in the era of deep ... [https://www.sciencedirect.com/science/article/pii/S1359644625001059]
- 4. Making sense of chemical space network shows signs ... [https://www.nature.com/articles/s41598-023-48107-3]
- 5. A View on Molecular Complexity from the GDB Chemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12381862/]
- 6. Traversing chemical space with active deep learning for ... [https://www.nature.com/articles/s43588-024-00697-2]
- 7. Active learning driven prioritisation of compounds from on ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00343h]
- 8. Navigating Chemical-Linguistic Sharing Space with ... [https://arxiv.org/html/2412.20888v1]
- 9. Active Learning in Machine Learning: Guide & Strategies ... [https://encord.com/blog/active-learning-machine-learning-guide/]
- 10. Leveraging active learning-enhanced machine ... [https://www.nature.com/articles/s41524-025-01827-8]
- 11. Understanding the Iterative Process (with Examples) [2025] [https://asana.com/resources/iterative-process]
- 12. A practical guide to Active Learning [https://www.avisi.nl/blog/a-practical-guide-to-active-learning]
- 13. Foundation Models for Discovery and Exploration in ... [https://arxiv.org/html/2510.18900v1]
- 14. The present state and challenges of active learning in drug ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644624001107]
- 15. Chemical Space Exploration with Active Learning and ... [https://www.lifescience.net/publications/1685531/chemical-space-exploration-with-active-learning-an/]
- 16. Advancing Drug Discovery with Enhanced Chemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12264933/]
- 17. Active Stacking-Deep Learning with Strategic Sampling for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12631473/]
- 18. The QDπ dataset, training data for drug-like molecules and ... [https://www.nature.com/articles/s41597-025-04972-3]
- 19. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 20. Chemical Space Exploration with Active Learning and ... [https://www.semanticscholar.org/paper/Chemical-Space-Exploration-with-Active-Learning-and-Khalak-Tresadern/e51d04db1c42a4b16ce8c745f99d916b062c4d49]
- 21. MvMRL: a multi-view molecular representation learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11200189/]
- 22. HLN-DDI: hierarchical molecular representation learning with ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06157-6]
- 23. Unified and explainable molecular representation learning ... [https://www.nature.com/articles/s41467-025-63730-6]
- 24. Exhaustive local chemical space exploration using a ... [https://www.nature.com/articles/s41467-024-51672-4]
- 25. Computational approaches streamlining drug discovery [https://www.nature.com/articles/s41586-023-05905-z]
- 26. Chemical Space Exploration with Active Learning and ... [https://pubmed.ncbi.nlm.nih.gov/36148968/]
- 27. Navigating Chemical Space: Multi-Level Bayesian ... [https://arxiv.org/html/2505.04169v1]
- 28. Hierarchy-boosted funnel learning for identifying ... [https://www.nature.com/articles/s41524-025-01583-9]
- 29. New AI model explores massive chemical space with minimal ... [https://pme.uchicago.edu/news/new-ai-model-explores-massive-chemical-space-minimal-data]
- 30. Best Practices for Alchemical Free Energy Calculations [Article ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8388617/]
- 31. Oracle Integration Cloud (OIC) [https://www.oracle.com/integration/application-integration/]
- 32. Rapid traversal of vast chemical space using machine ... [https://www.nature.com/articles/s43588-025-00777-x]
- 33. Ultra-large library screening with an evolutionary algorithm ... [https://www.nature.com/articles/s42004-025-01758-x]
- 34. An Accurate Quantitative Analysis Model for Martian-like ... [https://www.sciencedirect.com/science/article/abs/pii/S0950705125013723]
- 35. Exploring organic chemical space for materials discovery ... [https://www.nature.com/articles/s41467-025-65564-8]
- 36. Remapping the Chemical Space and the Pharmacological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11207054/]
- 37. Structure-based virtual screening of vast chemical space ... [https://www.sciencedirect.com/science/article/pii/S0959440X24000563]
- 38. A cAMP Biosensor-Based High-Throughput Screening Assay for... [https://pubmed.ncbi.nlm.nih.gov/25851033/]
- 39. Kinetic multiplex assay to assess biased signaling of ... [https://www.sciencedirect.com/science/article/pii/S1043661825004463]
- 40. Binding kinetics of ligands acting at GPCRs - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6406023/]
- 41. Knowledge-guided diffusion model for 3D ligand ... [https://www.nature.com/articles/s41467-025-57485-3]
- 42. Mechanistic insights into the identification of... | CoLab [https://colab.ws/articles/10.1016%2Fj.matchemphys.2025.131477]
- 43. GPCRdb in 2025: adding odorant receptors, data mapper ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11701689/]
- 44. Highly parallel optimisation of chemical reactions through ... [https://www.nature.com/articles/s41467-025-61803-0]
- 45. Active learning high coverage sets of complementary ... [https://pubs.rsc.org/en/content/articlelanding/2025/dd/d4dd00365a]
- 46. Optimizing drug design by merging generative AI with a ... [https://www.nature.com/articles/s42004-025-01635-7]
- 47. Active learning high coverage sets of complementary ... [https://www.sciencedirect.com/org/science/article/pii/S2635098X25000245]
- 48. Data-driven recommendation of agents, temperature, and ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04957a]
- 49. Deep learning for low-data drug discovery: Hurdles and ... [https://www.sciencedirect.com/science/article/pii/S0959440X24000459]
- 50. Active Learning Improves Ionization Efficiency Predictions and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12224155/]
- 51. Molecular property prediction in the ultra‐low data regime [https://www.nature.com/articles/s42004-025-01592-1]
- 52. A Quick Comparison of Experimental Results [https://www.chemedx.org/blog/quick-comparison-experimental-results]
- 53. An Integrated Method of Bayesian Optimization and D ... [https://www.mdpi.com/2227-9717/11/1/87]
- 54. Bayesian optimization with known experimental and ... [https://www.sciencedirect.com/org/science/article/abs/pii/S2635098X23000608]
- 55. An active machine learning approach for optimal design of ... [https://www.nature.com/articles/s41598-024-59100-9]
- 56. A Bayesian optimization approach for rapidly mapping ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8370405/]
- 57. Experimental Design Step by Step: A Practical Guide for ... [https://pubmed.ncbi.nlm.nih.gov/33258692/]
- 58. ACS Research Data Guidelines [https://researcher-resources.acs.org/publish/data_guidelines]
- 59. RLHF:从人类反馈中强化学习 [https://www.ultralytics.com/zh/glossary/reinforcement-learning-from-human-feedback-rlhf]
- 60. 什麼是RLHF?– 人類意見回饋強化學習介紹 [https://aws.amazon.com/tw/what-is/reinforcement-learning-from-human-feedback/]
- 61. Cutting-Edge Advancements in RLHF (2023–2025) [https://medium.com/foundation-models-deep-dive/cutting-edge-advancements-in-rlhf-2023-2025-fe814c770e88]
- 62. Traversing Chemical Space with Active Deep Learning: A ... [https://chemrxiv.org/engage/chemrxiv/article-details/65d8833ce9ebbb4db9098cb5]
- 63. FEP与3D-QSAR相结合的主动学习范式分析 [http://blog.molcalx.com.cn/2025/09/13/analysis-of-the-active-learning-paradigm-combining-fep-and-3d-qsar.html]
- 64. SynGFN: 面向可合成化学空间的生成式流网络分子生成方法 [https://cloud.tencent.com/developer/article/2590061]
- 65. RLHF and how real people are quietly training generative AI [https://www.thedrum.com/news/humans-the-loop-rlhf-and-how-real-people-are-quietly-training-generative-ai]
- 66. 主动学习优化的动态采样策略:提升化学化合物毒性预测的 ... [https://www.ebiotrade.com/newsf//2025-10/20251027105723027.htm]
- 67. [2503.22723] Zero-Shot LLMs in Human-in-the-Loop RL [https://arxiv.org/abs/2503.22723]
- 68. Machine learning in chemical reaction space [https://www.nature.com/articles/s41467-020-19267-x]
- 69. Rapid traversal of vast chemical space using machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12021657/]
- 70. Active learning streamlines development of high ... [https://www.nature.com/articles/s41467-024-50215-1]
- 71. Chemical Space Exploration with Active Learning and ... [https://figshare.com/articles/journal_contribution/Chemical_Space_Exploration_with_Active_Learning_and_Alchemical_Free_Energies/21196880]
- 72. Exploration of chemical space with partial labeled noisy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9063120/]
- 73. Active Learning Applications - Schrödinger [https://www.schrodinger.com/platform/products/active-learning-applications/]
- 74. opportunities for innovation in drug discovery - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12641420/]
- 75. The High-Throughput Screening Transformation in Modern ... [https://www.pharmasalmanac.com/articles/the-high-throughput-screening-transformation-in-modern-drug-development]
- 76. New AI model explores massive chemical space with minimal ... [https://climate.uchicago.edu/news/new-ai-model-explores-massive-chemical-space-with-minimal-data/]
- 77. Accelerated hit identification with target evaluation, deep ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00914-0]
- 78. Hit Identification and Optimization in Virtual Screening [https://pmc.ncbi.nlm.nih.gov/articles/PMC3772997/]
- 79. An Economic Framework to Prioritize Confirmatory Tests ... [https://www.sciencedirect.com/science/article/pii/S247255522207945X]
- 80. Binding Efficiency of Protein-Protein Complexes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3567247/]
- 81. Inhibitors supercharge kinase turnover through native ... [https://www.nature.com/articles/s41586-025-09763-9]
- 82. Prediction of protein–protein interaction based ... - BMC Biology [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-025-02359-9]
- 83. The present state and challenges of active learning in drug ... [https://pubmed.ncbi.nlm.nih.gov/38642700/]