Advanced SCF Convergence Strategies: Tackling Pathological Cases in Computational Chemistry and Drug Discovery
This article provides a comprehensive guide for researchers and drug development professionals on overcoming self-consistent field (SCF) convergence failures in computationally challenging molecular systems.
Advanced SCF Convergence Strategies: Tackling Pathological Cases in Computational Chemistry and Drug Discovery
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on overcoming self-consistent field (SCF) convergence failures in computationally challenging molecular systems. Pathological cases, such as open-shell transition metal complexes, systems with small HOMO-LUMO gaps, and conjugated radicals, are common hurdles in accurate electronic structure calculations for drug design. We explore the foundational causes of these failures, detail advanced methodological settings across major quantum chemistry packages (ORCA, Q-Chem, ADF), present a systematic troubleshooting protocol, and outline validation techniques to ensure physically meaningful results. By synthesizing current best practices and software-specific solutions, this guide aims to enhance the reliability and efficiency of computational workflows in biomedical research.
Understanding Pathological SCF Convergence: Why Standard Methods Fail
In computational chemistry, achieving Self-Consistent Field (SCF) convergence is a fundamental step for obtaining reliable electronic structure data. While modern SCF algorithms handle closed-shell organic molecules with relative ease, certain classes of compounds present significant challenges and are often classified as pathological cases for SCF convergence [1]. These problematic systems primarily include transition metal complexes, especially open-shell species, and specific organic systems such as conjugated radical anions with diffuse functions [1]. The inherent difficulties with these molecular systems stem from complex electronic structures, near-degeneracies, and the presence of multiple local minima on the electronic energy surface. Successfully converging these pathological cases requires specialized protocols and a deep understanding of SCF algorithms, as detailed in this application note.
Experimental Protocols for Pathological Systems
General SCF Convergence Assessment Protocol
Purpose: To diagnose and categorize the nature of SCF convergence failures. Methodology:
- Initial Calculation: Run a standard single-point energy calculation using density functional theory (DFT) with a moderate basis set (e.g., def2-SVP).
- Convergence Monitoring: Closely monitor the evolution of two key parameters:
- DeltaE (ΔE): The change in energy between SCF cycles.
- Orbital Gradients: The maximum (MaxP) and root-mean-square (RMSP) values of the orbital gradients [1].
- Failure Classification: Classify the convergence behavior based on the output:
- Slow Convergence: Steady but slow reduction of DeltaE and gradients.
- Oscillatory Behavior: Wild fluctuations in energy or densities between cycles.
- True Stagnation: Minimal to no progress toward convergence after initial iterations.
Initial SCF Diagnosis
Specialized Protocol for Transition Metal Complexes
Purpose: To achieve SCF convergence for open-shell transition metal compounds, which are notorious for pathological behavior due to their dense electronic structure and near-degenerate states [1].
Methodology:
- Initial Attempt with Enhanced Damping:
- Algorithm Selection for Stubborn Cases:
- Employ the
!KDIISalgorithm, sometimes combined with!SOSCFfor accelerated convergence once a stable path is found [1]. - Caution: For open-shell systems, the SOSCF algorithm is turned off by default and can be unstable. If used, delay its activation by setting
SOSCFStartto a more stringent value (e.g.,0.00033) [1].
- Employ the
- Utilization of Second-Order Convergers:
- Advanced Troubleshooting for Pathological Cases:
- For extremely difficult systems (e.g., metal clusters), implement a high-cost, high-reliability protocol:
- Use
!SlowConv. - Increase
MaxIterto 1500. - Expand the DIIS extrapolation space with
DIISMaxEq 15-40. - Reduce numerical noise by setting
directresetfreq 1to rebuild the Fock matrix every iteration [1].
- Use
- For extremely difficult systems (e.g., metal clusters), implement a high-cost, high-reliability protocol:
Specialized Protocol for Conjugated Radical Anions
Purpose: To converge the SCF for conjugated radical anions, where the combination of a diffuse electron cloud and an open-shell structure leads to convergence pathologies [1].
Methodology:
- Fock Matrix Rebuild: Set
directresetfreq 1to ensure a full rebuild of the Fock matrix in each iteration, mitigating convergence issues caused by numerical noise in systems with diffuse functions [1]. - Early SOSCF Activation: Configure the SCF block to initiate the SOSCF algorithm earlier than default to accelerate convergence:
soscfmaxit 12[1].
Data Presentation and Analysis
Quantitative SCF Settings for Pathological Cases
The following table summarizes the key SCF parameters and their recommended values for different pathological scenarios, serving as a quick-reference guide for researchers.
Table 1: Recommended SCF Settings for Pathological Convergence Cases
| Pathological Case | Key ORCA Keywords | SCF Block Parameters | Typical Value Ranges | Primary Function |
|---|---|---|---|---|
| General Difficult Systems | !SlowConv, !VerySlowConv |
Shift 0.1 ErrOff 0.1 |
N/A | Applies damping to control initial oscillations [1]. |
| Stubborn Transition Metals | !KDIIS, !SOSCF |
SOSCFStart 0.00033 |
N/A | Uses alternative algorithm; delays SOSCF for stability [1]. |
| Pathological Cases (e.g., Fe-S Clusters) | !SlowConv |
MaxIter 1500, DIISMaxEq 15, directresetfreq 1 |
15-40 for DIISMaxEq | Maximizes iteration count, DIIS space, and reduces numerical noise [1]. |
| Conjugated Radical Anions | (None specified) | directresetfreq 1, soscfmaxit 12 |
N/A | Ensures exact Fock builds and accelerates convergence [1]. |
The Scientist's Toolkit: Essential Research Reagents and Computational Methods
This toolkit outlines critical computational strategies and their specific applications in resolving SCF convergence pathologies.
Table 2: Essential Computational Toolkit for Resolving SCF Pathologies
| Tool / Reagent | Function / Application | Key Considerations |
|---|---|---|
| Initial Orbital Guess (PModel) | Generates starting orbitals for the SCF procedure. | Default in ORCA; sufficient for most non-pathological cases [1]. |
| Alternative Guesses (HCore, PAtom) | Provides a more robust starting point when the default guess fails. | PAtom uses atomic densities; can be more stable for metals [1]. |
| MORead | Reads orbitals from a previously converged, simpler calculation (e.g., BP86/def2-SVP). | Excellent for providing a stable initial guess from a related, well-behaved system [1]. |
| Damping (!SlowConv) | Suppresses large oscillations in the early SCF iterations. | First-line response for oscillating or slowly converging systems [1]. |
| Level-Shifting (%scf Shift) | Shifts orbital energies to stabilize the SCF process. | An alternative to damping for preventing variational collapses [1]. |
| DIIS (Direct Inversion in Iterative Subspace) | Extrapolates Fock matrices to accelerate convergence. | Default algorithm; increasing DIISMaxEq can help difficult cases [1]. |
| SOSCF (Second Order SCF) | Uses Newton-Raphson method for fast convergence near the solution. | Not default for open-shell; requires careful activation [1]. |
| TRAH (Trust Radius Augmented Hessian) | A robust second-order convergence algorithm. | Automatically activates in ORCA 5.0+ if DIIS struggles; more reliable but slower [1]. |
Integrated Workflow for Managing Pathological Convergence
The following diagram synthesizes the protocols and tools into a cohesive decision-making workflow for tackling the most challenging SCF cases, particularly focusing on transition metal systems.
Comprehensive Workflow for Pathological Transition Metal Complexes
Self-Consistent Field (SCF) methods form the computational backbone for electronic structure calculations in quantum chemistry and materials science, enabling the modeling of molecular and solid-state systems. Despite their widespread use, SCF procedures frequently encounter convergence failures, particularly when dealing with systems exhibiting small HOMO-LUMO gaps or orbital degeneracy. These failures stem from fundamental physical and mathematical challenges inherent in the SCF iterative process. This application note examines the theoretical roots of these convergence pathologies, provides diagnostic protocols for their identification, and details robust computational strategies to overcome them, with a specific focus on their implications for research in drug development and materials design.
Physical and Numerical Origins of SCF Failures
The SCF convergence process is fundamentally an iterative optimization problem that can exhibit chaotic behavior [2]. When the energy separation between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) becomes small, the electronic structure becomes susceptible to numerical instabilities. These instabilities manifest primarily through two physical mechanisms: occupation number switching and charge sloshing [3].
Orbital Degeneracy and Near-Degeneracy Effects
Orbital degeneracy occurs when two or more molecular orbitals possess identical or nearly identical energies. In such scenarios, even minute numerical noise can cause electrons to switch between degenerate or near-degenerate orbitals during SCF iterations. This creates an oscillatory pattern where:
- At iteration N, electrons occupy orbital ψ1 while ψ2 remains vacant
- At iteration N+1, orbital energies shift, causing electrons to transfer from ψ1 to ψ2
- At iteration N+2, the electron density redistribution reverses this occupation pattern
This cyclic behavior prevents the electron density from stabilizing, resulting in non-convergence [3]. Systems with incorrect symmetry specifications can exacerbate this problem, sometimes leading to exactly zero HOMO-LUMO gaps that make convergence virtually impossible [3].
Charge Sloshing in Systems with High Polarizability
The polarizability of a molecular system is inversely proportional to its HOMO-LUMO gap [3]. As this gap shrinks, the system becomes increasingly polarizable, meaning that small errors in the Kohn-Sham potential can produce large distortions in the electron density. When these distorted densities generate even more erroneous potentials in subsequent iterations, the process enters a positive feedback loop of escalating density oscillations—a phenomenon known as "charge sloshing" [3] [4]. This manifests as long-wavelength oscillations of the output charge density during SCF iterations, typically with energy oscillations of moderate amplitude (10⁻⁴-10⁻² Hartree) while maintaining qualitatively correct orbital occupation patterns [3].
Table 1: Characteristic Signatures of SCF Convergence Failures
| Failure Mechanism | Energy Oscillation Amplitude | Orbital Occupation Pattern | Density Convergence |
|---|---|---|---|
| Occupation Switching | Large (10⁻⁴ - 1 Hartree) | Clearly wrong, oscillating | Poor |
| Charge Sloshing | Moderate (~10⁻⁴ Hartree) | Qualitatively correct | Poor |
| Numerical Noise | Very small (<10⁻⁴ Hartree) | Qualitatively correct | Stalled |
Diagnostic Protocols and Identification
Identifying the specific type of SCF convergence failure requires systematic analysis of the SCF output. The following diagnostic protocol enables researchers to distinguish between different failure mechanisms.
SCF Output Analysis Protocol
Monitor Energy Convergence: Track the total energy difference between successive SCF iterations. Oscillations with large amplitudes (10⁻⁴ - 1 Hartree) typically indicate occupation number switching, while smaller oscillations suggest charge sloshing or numerical noise [3].
Analyze Orbital Occupations: Examine the final orbital occupation pattern printed at the end of the SCF calculation. Compare this with chemical intuition about the expected electronic structure. Clearly incorrect occupation patterns indicate fundamental occupation switching problems [3].
Check Density Matrix Convergence: Assess the root-mean-square change in the density matrix (RMSDP). In pathological cases, the energy may appear converged while the density matrix shows significant oscillations (>10⁻²) [5].
Evaluate HOMO-LUMO Gap: Calculate the HOMO-LUMO gap from initial iterations. Gaps below ~0.1 eV often presage convergence difficulties and may require preemptive stabilization measures [3].
Test Basis Set Dependence: Repeat calculations with smaller basis sets. Convergence failures that persist across different basis sets indicate physical rather than numerical problems [5].
The following workflow illustrates the diagnostic process for identifying SCF convergence pathologies:
Case Study: Pathological Geometry Analysis
A concrete example demonstrates these diagnostics in practice. For a system with atoms at pathological separations, the SCF output may show:
Here, the energy has converged to numerical precision (ΔE ~ 10⁻¹³ Hartree), but the density matrix remains far from convergence (RMSDP = 1.01356×10⁻²), indicating a severe charge sloshing problem [5].
Computational Methodologies for Pathological Cases
Advanced SCF Convergence Algorithms
For systems with small HOMO-LUMO gaps or orbital degeneracy, standard DIIS (Direct Inversion in the Iterative Subspace) algorithms often fail, necessitating specialized approaches:
Second-Order SCF (SOSCF): This algorithm uses orbital Hessian information to achieve more reliable convergence, though at increased computational cost per iteration. SOSCF is particularly valuable for open-shell systems and metal complexes where conventional methods fail [6].
Maximum Overlap Method (MOM) and Variants: Designed for ΔSCF calculations targeting excited states, MOM maintains convergence toward a specific electronic configuration by maximizing overlap with an initial reference configuration [7]. The Initial MOM (IMOM) preserves the initial reference state throughout the SCF procedure, while Projected MOM (PMOM) offers alternative overlap metrics [7].
Damping and Level Shifting: Applying a damping percentage (e.g., 20%) reduces step sizes in the SCF iteration, while level shifting (e.g., 0.1 Hartree) artificially increases the energy of unoccupied orbitals, effectively widening the HOMO-LUMO gap during early iterations [5] [2].
Table 2: Research Reagent Solutions for SCF Convergence Problems
| Method | Key Function | Typical Parameters | Applicable Failure Mode |
|---|---|---|---|
| SOSCF | Uses orbital Hessian for stable convergence | Automatic switch upon failure | All pathological cases, especially metal complexes |
| MOM/IMOM | Maintains target orbital occupation | ALPHACONF 0,1 for HOMO→LUMO excitation |
Occupation switching, ΔSCF calculations |
| Damping | Reduces step size between iterations | DAMPING_PERCENTAGE 20 |
Charge sloshing, oscillation |
| Level Shifting | Artificially increases virtual orbital energies | LEVEL_SHIFT 0.1 (Hartree) |
Small HOMO-LUMO gaps |
| ADIIS | Alternative to DIIS for difficult cases | Hybrid DIIS/ADIIS strategy | Stagnating convergence |
| Smearing | Introduces fractional occupations | Fermi-Dirac, 0.2-0.5 eV | Metallic systems, near-degeneracy |
ΔSCF Protocol for Excited States
The ΔSCF approach enables calculation of excited-state properties by converging to non-Aufbau orbital configurations. The following protocol outlines its implementation:
Generate Ground-State Orbitals: First, converge a standard ground-state calculation and save the orbital coefficients.
Define Target Configuration: Specify the desired orbital occupation using appropriate keywords (e.g.,
ALPHACONF 0,1for exciting one electron from HOMO to LUMO) [7].Configure SCF Settings:
- Set
DELTASCFin the main input - Select
UHFfor open-shell singlet states orRHFfor doubly-excited states - Use
L-SR1Hessian updates instead of defaultL-BFGSto converge to saddle points - Enable
KEEPINITIALREF TRUEto maintain the initial reference state [7]
- Set
Monitor Spin Contamination: For open-shell singlets, check the 〈S²〉 value. Significant deviation from the ideal value (0.0 for singlets) indicates spin contamination, though the charge distribution may remain physically reasonable [7].
System-Specific Stabilization Techniques
Different system characteristics demand tailored approaches:
Metallic Systems with Small Gaps: Apply Fermi-Dirac or Gaussian smearing (0.2-0.5 eV) to stabilize fractional occupation changes [4].
Antiferromagnetic Systems: Use reduced mixing parameters (e.g.,
AMIX = 0.01,BMIX = 1e-5) for both charge and spin density mixing [4].Elongated Systems: For cells with large aspect ratios, employ "local-TF" mixing or significantly reduce mixing parameters (
beta=0.01) to address ill-conditioning [4].Numerical Grid Sensitivity: Use dense integration grids (≥99 radial, 590 angular points) to minimize numerical noise, particularly for meta-GGA functionals [2].
SCF convergence failures stemming from orbital degeneracy and small HOMO-LUMO gaps represent fundamental challenges in electronic structure theory. Through systematic diagnosis of the failure mode and application of specialized algorithms like SOSCF, MOM, and controlled density mixing, researchers can overcome these pathologies. The protocols outlined in this application note provide a structured approach to addressing these challenges, enabling reliable computation of electronic properties for chemically complex systems relevant to drug development and materials design. As functional complexity increases in modern DFT, robust SCF convergence strategies become increasingly essential for producing physically meaningful results across diverse chemical spaces.
The Self-Consistent Field (SCF) procedure is the fundamental method for solving the electronic structure problem in quantum chemistry. However, achieving convergence remains a significant challenge for specific classes of molecules and electronic structures. Pathological convergence cases frequently arise from three common culprits: open-shell configurations, significant antiferromagnetic exchange coupling, and the use of diffuse basis functions. These systems often exhibit small HOMO-LUMO gaps, strong electron correlation effects, and numerical instabilities that impede the SCF process [1] [8].
This application note provides a structured framework for identifying and resolving these challenging SCF convergence failures. It synthesizes current knowledge and practical protocols into actionable strategies, including diagnostic workflows, optimized parameter settings, and detailed experimental methodologies tailored for computational researchers and drug development scientists engaged in molecular modeling of complex systems.
The Scientist's Toolkit: Research Reagent Solutions
Table 1: Essential computational tools and parameters for tackling pathological SCF convergence.
| Tool Category | Specific Example/Setting | Primary Function |
|---|---|---|
| SCF Convergers | DIIS, KDIIS, TRAH, SOSCF | Accelerate or stabilize SCF convergence via Fock matrix extrapolation or second-order methods [1]. |
| Initial Guess Strategies | PAtom, Hückel (HCore), MORead |
Generate improved starting orbitals, bypassing inadequate default guesses [1]. |
| Damping/Levelshift | SlowConv, Shift 0.1, ErrOff 0.1 |
Suppress oscillatory SCF behavior by mixing old/new densities or shifting virtual orbitals [1]. |
| Advanced Keywords | DirectResetFreq 1, DIISMaxEq 40 |
Reduce numerical noise by frequent Fock rebuilds and expand DIIS subspace for difficult cases [1]. |
| Basis Sets | ma-def2-SVP, aug-cc-pVTZ |
Diffuse functions for accurate anion/anion interaction description; may cause linear dependence [1] [8]. |
Quantitative Data and Convergence Criteria
SCF Convergence Thresholds
Table 2: Standard and system-dependent SCF convergence criteria. The default behavior in ORCA distinguishes between levels of convergence, and criteria can vary with numerical settings [1] [9].
| Convergence Level | DeltaE (Hartree) | Max Density | RMS Density | Post-SCF Action (Single Point) |
|---|---|---|---|---|
| Complete Convergence | Below Set Criterion | Below Set Criterion | Below Set Criterion | Calculation proceeds normally. |
| Near Convergence | < 3.0e-3 | < 1.0e-2 | < 1.0e-3 | ORCA stops with a warning; energy marked as not fully converged [1]. |
| No Convergence | Above Threshold | Above Threshold | Above Threshold | ORCA stops; user intervention required. |
System-Specific SCF Settings for Pathological Cases
Table 3: Optimized SCF parameters for different classes of problematic molecules. These settings represent a starting point for troubleshooting specific convergence failures [1].
| System Type | Key SCF Settings | Expected Performance Impact |
|---|---|---|
| Open-Shell Transition Metals | !SlowConv SOSCF, SOSCFStart 0.00033, Shift 0.1, ErrOff 0.1 |
Moderate slowdown due to damping and delayed SOSCF activation. |
| Pathological Clusters/Radicals | !VerySlowConv, DIISMaxEq 15-40, DirectResetFreq 1, MaxIter 1500 |
Significant slowdown due to expensive Fock rebuilds and large DIIS space. |
| Conjugated Radical Anions (Diffuse Functions) | DirectResetFreq 1, SOSCFStart 0.00033 |
Moderate slowdown due to full Fock rebuilds but enables convergence [1]. |
Experimental Protocols
Protocol 1: Systematic SCF Convergence Workflow for Pathological Geometries
Purpose: To provide a step-by-step methodology for diagnosing and remedying SCF convergence failures in complex molecular systems, such as open-shell transition metal complexes or molecules with pathological geometries [1] [5].
Required Inputs: Molecular geometry (e.g., XYZ coordinates), charge, multiplicity, and basis set.
Procedure:
- Initial Diagnosis:
- Run a single-point energy calculation with default SCF settings (
!NormalSCF). - Examine the output for the convergence trajectory (e.g., oscillating vs. stalled energy/density) and final error messages.
- Critical Check: Verify the reasonableness of the input geometry. Pathological geometries, such as atoms at extreme separations, are a common root cause and may require geometric preprocessing before SCF troubleshooting [5].
- Run a single-point energy calculation with default SCF settings (
Tier 1 Interventions (Mild): Apply if the SCF shows signs of convergence but fails within the default iteration limit.
- Increase the maximum number of SCF cycles:
%scf MaxIter 500 end. - Use a
MOReadguess from a pre-converged calculation with a simpler method (e.g., BP86/def2-SVP):!MORead "%moinp "prev_calc.gbw""[1].
- Increase the maximum number of SCF cycles:
Tier 2 Interventions (Moderate): Apply for strong oscillations or slow convergence.
- Activate damping algorithms:
!SlowConvor!VerySlowConv. - Introduce level-shifting:
%scf Shift 0.1 ErrOff 0.1 end. - For open-shell systems, try the KDIIS algorithm:
!KDIIS[1].
- Activate damping algorithms:
Tier 3 Interventions (Aggressive): For truly intractable cases, such as metal clusters [1].
- Significantly expand the DIIS subspace and force frequent Fock matrix rebuilds:
- If the TRAH algorithm is activated but is slow, adjust its parameters or disable it with
!NoTrahand rely on the above DIIS settings [1].
Final Validation:
- Once an SCF setting enables convergence, always confirm that the final single-point energy is marked as fully converged.
- For geometry optimizations, ensure that the SCF remains stable across multiple optimization cycles.
Diagram 1: SCF Convergence Workflow
Protocol 2: Electronic Structure Analysis of an Open-Shell Biradicaloid Complex
Purpose: To characterize the electronic structure of an oligothiophene-bridged bis(semiquinone) complex (SQ-Th2-SQ), which exhibits significant open-shell character and antiferromagnetic coupling [8]. This protocol outlines the combined use of computational and experimental techniques.
System: SQ-Thn-SQ (n = 0-3) complexes, where SQ is a Zn(II)-bound semiquinone radical (S = ½) [8].
Computational and Experimental Methodology:
- Synthesis and Crystallization:
- Synthesize the target
SQ-Thn-SQcomplexes via coordination chemistry of the bridging ligand and the TpCum,MeZnII metal complex. - Grow single crystals suitable for X-ray diffraction analysis.
- Synthesize the target
Structural Analysis (Quinoidal vs. Aromatic Character):
- Collect X-ray crystallographic data.
- Analyze the bond lengths within the oligothiophene bridge. Calculate the Bridge Bond Deviation value (Σ|Δi|) by comparing bond lengths to reference aromatic systems.
- Interpretation: A quinoidal bonding pattern indicates a more closed-shell ground state, while aromatic bond lengths are characteristic of an open-shell biradical [8].
Magnetic Susceptibility Measurements:
- Perform variable-temperature (VT) magnetic susceptibility measurements (e.g., using a SQUID magnetometer).
- Fit the magnetic susceptibility (χ) vs. temperature (T) data to the Bleaney-Bowers equation for a biradical model to determine the singlet-triplet energy gap, quantified by the exchange coupling constant, J [8].
Spectroscopic Characterization:
- VT Electronic Absorption Spectroscopy: Identify low-energy optical transitions that correspond to the population of open-shell excited states.
- VT Electron Paramagnetic Resonance (EPR) Spectroscopy: Detect and characterize paramagnetic triplet states at elevated temperatures, providing an independent measure of the zero-field splitting (D) and J coupling [8].
Computational Modeling:
- Perform broken-symmetry DFT calculations to estimate the exchange coupling constant J and map the spin density distribution across the molecule.
- Use a simplified 4-electron, 3-orbital model to interpret the configurational mixing between closed-shell quinoid and open-shell biradical states based on molecular symmetry [8].
Diagram 2: Biradicaloid Characterization
Discussion and Analysis
The convergence pathologies discussed are intrinsically linked to the electronic structure of the system. In open-shell biradicaloids like the SQ-Thn-SQ series, the energy gap between the closed-shell quinoid configuration and the open-shell biradical configuration becomes very small [8]. This results in a near-degeneracy that severely challenges the SCF procedure. The presence of significant antiferromagnetic coupling, as measured by a large negative J value (e.g., -279 cm⁻¹ for SQ-Th2-SQ), confirms a low-lying triplet state that can be thermally populated, further complicating the convergence of a single-determinant method [8].
The use of diffuse basis functions exacerbates these issues by introducing numerical linear dependencies and increasing the condition number of the overlap matrix, which is particularly problematic when modeling conjugated radical anions or using large basis sets like aug-cc-pVTZ [1]. The recommended strategy of setting DirectResetFreq 1 for such systems works by minimizing the accumulation of numerical noise in the constructed Fock matrices, which is a critical source of instability in these delicate cases [1].
Successfully converging the SCF for pathological cases requires a systematic approach that combines robust computational protocols with a deep understanding of underlying electronic structure principles. The strategies and experimental frameworks detailed herein—ranging from simple parameter adjustments to advanced multi-method characterization—provide a proven pathway for tackling even the most challenging open-shell, antiferromagnetically coupled, and diffuse-function-dependent systems. Mastery of these techniques is essential for advancing research in catalysis, material science, and drug development where such complex electronic structures are commonplace.
Self-Consistent Field (SCF) methods, foundational to both Hartree-Fock theory and Kohn-Sham Density Functional Theory, represent an iterative process to solve the electronic structure problem. In this procedure, the Fock matrix must be made consistent with the density matrix it helps determine, leading to an iterative cycle that ideally converges to a stable solution. However, pathological convergence cases present significant challenges in computational chemistry, particularly for researchers investigating complex molecular systems in drug development. Such cases are frequently encountered with open-shell transition metal compounds, systems with very small HOMO-LUMO gaps, and conjugated radical anions with diffuse functions. Modern quantum chemistry codes have developed sophisticated algorithms to address these issues, yet understanding and diagnosing the specific patterns of convergence failure remains crucial for obtaining physically meaningful results. The behavior of the SCF process, when it fails to converge, provides critical diagnostic information that guides the selection of appropriate remediation strategies. This application note provides a systematic framework for analyzing SCF iteration output and implementing solutions for pathological cases.
Recognizing Convergence Patterns from Output
The evolution of key quantities across SCF iterations reveals specific patterns that identify the underlying cause of convergence difficulties. Diagnosing the problem correctly is the first step toward applying an effective solution.
Quantitative Convergence Criteria
SCF convergence is typically assessed through multiple metrics that should all approach zero as the calculation reaches self-consistency. The most common criteria and their interpretations are summarized in Table 1.
Table 1: Key SCF Convergence Metrics and Their Interpretation
| Metric | Mathematical Form | Physical Significance | Convergence Threshold |
|---|---|---|---|
| DeltaE | ΔE = |Eₙ - Eₙ₋₁| | Change in total energy between iterations | Typically < 10⁻⁶ to 10⁻⁸ a.u. [1] |
| Max Density/Potential Error | Max |PS - SP| | Maximum element of the commutator [F,P] | Typically < 10⁻⁴ to 10⁻⁵ [1] [10] |
| RMS Density/Potential Error | RMS |PS - SP| | Root-mean-square of the commutator [F,P] | Typically < 10⁻⁵ [1] |
| Orbital Gradient | ∂E/∂C | Gradient of energy with respect to orbital coefficients | Should approach zero [1] [11] |
Most quantum chemistry programs define distinct convergence states. For instance, ORCA distinguishes between "complete SCF convergence," "near SCF convergence" (deltaE < 3e-3; MaxP < 1e-2; RMSP < 1e-3), and "no SCF convergence" [1]. Near convergence outcomes still permit geometry optimizations to continue but trigger warnings in single-point calculations.
Oscillation Patterns and Their Diagnosis
Oscillations in the SCF energy or error metrics represent a common pathological pattern where values alternate between two or more states without approaching convergence.
Table 2: Characteristic Oscillation Patterns and Their Meanings
| Oscillation Pattern | Typical Characteristics | Common System Types | Underlying Cause |
|---|---|---|---|
| Two-State Limit Cycle | Energy and wavefunction alternate between two distinct states [12] | Systems with near-degenerate orbitals | Non-linear iterated functional mapping converging to a limit cycle rather than a fixed point |
| Damped Oscillations | Oscillations with decreasing amplitude | Systems initially far from solution | Overly aggressive convergence acceleration |
| Growing Oscillations | Increasing amplitude with iteration | Unphysical descriptions or incorrect multiplicity | Divergence indicating fundamental issues with the calculation setup |
Oscillatory behavior often occurs when the SCF procedure "jumps back and forth between two answers" [12], particularly in systems with small HOMO-LUMO gaps where the orbital energy spectrum presents near-degenerate states. This behavior represents convergence to a two-state limit cycle, a fundamental non-linear phenomenon in the functional mapping of the SCF process.
Systematic Protocol for Diagnosing SCF Convergence Issues
The following step-by-step protocol provides a structured approach to diagnosing SCF convergence problems based on iteration output analysis.
Initial Assessment and Artifact Elimination
- Verify Molecular Geometry: Inspect bond lengths, angles, and coordination geometries for physical realism. High-energy geometries or unphysical atomic arrangements frequently cause convergence failure [13].
- Confirm Electronic State: Validate that the specified spin multiplicity (singlet, doublet, triplet, etc.) and charge correspond to the intended electronic state of the system. Incorrect multiplicity represents a common source of SCF instability [13].
- Check Basis Set Appropriateness: Ensure the selected basis set does not exhibit linear dependencies, particularly when using diffuse functions or for metal-containing systems [1].
- Examine Initial Iterations: Review the first 5-10 SCF iterations. Wild oscillations appearing immediately often indicate an poor initial guess or fundamental issues with the molecular system [1].
Output Analysis and Pattern Recognition
- Plot Convergence Metrics: Generate plots of DeltaE, Max Density Error, and RMS Density Error versus iteration number.
- Classify the Pattern: Categorize the behavior as:
- Smooth but slow convergence: Steady decrease in errors without oscillation
- Oscillatory: Regular alternation between values
- Divergent: Steady increase in errors
- Stagnant: Little to no change after initial improvements
- Identify the Onset: Note the iteration where problematic behavior begins. Late-onset oscillations (after 10-20 iterations) suggest different remedies than early-onset issues.
- Check for Near-Convergence: Determine if the calculation meets "near convergence" criteria, which may be acceptable for certain computational goals [1].
Advanced Diagnostics
- Stability Analysis: Perform a stability check on the converged (or nearly converged) wavefunction to determine if it represents a true minimum or a saddle point [11].
- Orbital Inspection: Examine the molecular orbitals, particularly the HOMO-LUMO region, for unusual degeneracies or spatial characteristics.
- Density Difference Analysis: Plot the difference between input and output densities to identify regions of strong fluctuation.
The following workflow diagram illustrates the logical decision process for diagnosing SCF convergence issues:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Reagents for SCF Convergence Troubleshooting
| Reagent/Solution | Function | Application Context | Key Parameters |
|---|---|---|---|
| DIIS Extrapolation | Accelerates convergence by extrapolating from previous Fock matrices [10] | Default algorithm for most systems; effective for stable convergence | DIISSUBSPACESIZE (default 5-15, increase to 25-40 for difficult cases) [1] |
| Damping | Stabilizes oscillations by mixing old and new Fock matrices | Early-stage oscillations; fluctuating iterations | DAMP factor (0.2-0.5); applied in initial cycles [11] [14] |
| Level Shifting | Increases HOMO-LUMO gap by raising virtual orbital energies | Small-gap systems; oscillatory convergence | LEVEL_SHIFT (0.001-0.5 Ha); enables convergence but affects virtual orbitals [11] [13] |
| SOSCF | Second-order convergence using orbital Hessian | When near convergence but trailing off; reduces iterations | SOSCFStart (default 0.0033, reduce to 0.00033 for early activation) [1] |
| TRAH | Trust-region augmented Hessian method | Robust but expensive alternative when DIIS fails | AutoTRAHTOl (default 1.125); AutoTRAHIter (default 20) [1] |
| Geometric Direct Minimization (GDM) | Robust minimization accounting for orbital rotation space geometry | DIIS failure; restricted open-shell systems | SCF_ALGORITHM = GDM [10] |
| Electron Smearing | Fractional occupancies to overcome near-degeneracies | Metallic systems; small HOMO-LUMO gaps | Smearing width (keep as low as possible) [11] [13] |
| Initial Guess Alternatives | Improved starting point for SCF iterations | Default guess fails; transition metal systems | atom, huckel, read from checkpoint file [11] |
Experimental Protocols for Pathological Cases
Protocol for Oscillating Transition Metal Complexes
Transition metal complexes, particularly open-shell systems, represent particularly challenging cases for SCF convergence. The following protocol has been specifically optimized for these systems:
Initial Setup:
Algorithm Selection:
- Start with
!SlowConvkeyword or equivalent, which applies damping parameters appropriate for difficult systems [1]. - For ORCA, implement combined DIIS and SOSCF approach:
! KDIIS SOSCFwith delayed SOSCF start:%scf SOSCFStart 0.00033 end[1]. - For Q-Chem, use
SCF_ALGORITHM = DIIS_GDMto benefit from initial DIIS acceleration followed by robust GDM convergence [10].
- Start with
Parameter Optimization:
Advanced Techniques:
Protocol for Systems with Small HOMO-LUMO Gaps
Systems with vanishing HOMO-LUMO gaps, such as conjugated polyradicals, metal clusters, and systems with diffuse basis functions, require specialized approaches:
Initial Steps:
- Employ electron smearing with a small width (0.001-0.01 Ha) to facilitate initial convergence [11] [13].
- Use density matrix mixing with low mixing parameters (0.05-0.15) rather than DIIS in initial iterations [13].
- Implement the
huckelinitial guess or similar parameter-free methods designed for difficult cases [11].
Convergence Algorithm:
Specialized Settings:
- For conjugated radical anions with diffuse functions, increase Fock matrix rebuild frequency:
directresetfreq 1and activate SOSCF early [1]. - Implement fractional occupations or thermal smearing to stabilize the density matrix [11].
- Use
DIIS_SEPARATE_ERRVEC = TRUEin Q-Chem if alpha and beta error cancellation is suspected [10].
- For conjugated radical anions with diffuse functions, increase Fock matrix rebuild frequency:
Protocol for Oscillation Analysis and Remediation
When confronting clear oscillatory behavior, the following targeted protocol applies:
Characterization:
- Plot energy difference (ΔE) versus iteration number to identify oscillation period and amplitude.
- Determine if oscillations involve the entire electron density or specific molecular regions through density difference plots.
- Check for oscillation between different orbital occupancy patterns (e.g., different orbital ordering).
Initial Intervention:
Advanced Interventions:
- Implement the Maximum Overlap Method (MOM) to maintain consistent orbital occupancy across iterations [10].
- Switch to a direct minimization algorithm (GDM or DM) bypassing DIIS entirely [10].
- Utilize the Augmented Roothaan-Hall (ARH) method or similar conjugate-gradient approaches available in codes like ADF [13].
Validation:
- After convergence, perform wavefunction stability analysis to ensure a true minimum has been located [11].
- Verify that the solution is physically reasonable through examination of molecular orbitals, spin densities, and population analysis.
Diagnosing SCF convergence problems requires systematic analysis of iteration output to identify characteristic patterns, particularly oscillatory behavior. Through careful application of the protocols and tools outlined in this application note, researchers can effectively address even pathological convergence cases in complex molecular systems relevant to drug development. The key to success lies in methodical pattern recognition followed by targeted application of algorithmic solutions from the computational toolkit. Future developments in SCF algorithms, particularly robust second-order methods and improved initial guesses, continue to expand the range of tractable systems while reducing the need for manual intervention.
The Critical Role of Initial Guess Quality in Difficult Calculations
In quantum chemistry calculations, the Self-Consistent Field (SCF) method is an iterative procedure used to solve the electronic structure of molecules in both Hartree-Fock and Kohn-Sham Density Functional Theory. The quality of the initial guess for the molecular orbitals significantly impacts the convergence behavior of these calculations. A poor initial guess can lead to slow convergence, convergence to incorrect electronic states, or complete SCF failure, particularly for pathological systems such as open-shell transition metal complexes, species with multireference character, or molecules at unphysical geometries. This application note details the critical importance of initial guess selection and provides structured protocols for researchers dealing with challenging convergence cases, framed within broader research on SCF block settings for pathological convergence.
Quantitative Assessment of Initial Guess Methods
Performance Comparison of Common Initial Guess Methods
A systematic assessment of initial guess methods performed on 259 molecules ranging from first to fourth periods revealed significant performance differences across methods. The study projected guess orbitals onto precomputed, converged SCF solutions in single- to triple-ζ basis sets to evaluate accuracy [15].
Table 1: Performance Characteristics of Initial Guess Methods
| Method | Description | Average Performance | Key Strengths | Implementation Considerations |
|---|---|---|---|---|
| SAP (Superposition of Atomic Potentials) | Constructs guess from summed atomic potentials | Best performance on average [15] | Simple yet efficient; easily implementable in real-space calculations [15] | Resembles parameter-free extended Hückel method [15] |
| SAD (Superposition of Atomic Densities) | Sums spherically-averaged atomic densities to form trial density matrix | Good performance | Popular choice; superior to core Hamiltonian or GWH guesses [16] | Not idempotent; requires at least two SCF iterations [16] |
| Extended Hückel | Uses minimal basis (STO-3G) extended Hückel calculation | Good alternative with less scatter in accuracy [15] | Parameter-free variant resembles SAP method [15] | Easy to implement on existing SAD infrastructure [15] |
| GWH (Generalized Wolfsberg-Helmholtz) | Combines overlap matrix and core Hamiltonian diagonal elements [16] | Satisfactory for small molecules/basis sets [16] | Simple construction | Performance degrades with increased molecule and basis set size [16] |
| Core Hamiltonian | Diagonalizes core Hamiltonian matrix | Simplest approach [17] | Often a "disaster"; produces orbitals that are too compact [17] | Works best with small basis sets; degrades severely with larger systems [16] |
Specialized Guess Methods for Challenging Systems
Table 2: Specialized Initial Guess Approaches for Pathological Cases
| Method | Application Context | Protocol | Expected Outcome |
|---|---|---|---|
| Fragment MO (FRAGMO) | Multi-component systems; fragment-based calculations [16] | Superimpose converged fragment molecular orbitals | Preserves local electronic structure of components |
| Basis Set Projection (BASIS2) | Calculations requiring large basis sets [16] | 1. Perform DFT calculation in small basis set2. Project density matrix to large basis3. Begin target SCF calculation | Reduced number of iterations in target basis |
| Oxidized/Reduced State Convergence | Open-shell systems with convergence difficulties [1] | 1. Converge closed-shell cation/anion2. Read orbitals into neutral system calculation | Breaking symmetry constraints of original system |
| Orbital Rotation and Swapping | Targeting specific electronic states; breaking symmetry [16] [17] | Use $occupied or $swap_occupied_virtual keywords to modify occupancy |
Guides SCF to desired local minimum in wavefunction space |
Initial Guess Selection Workflow
The following decision diagram provides a systematic workflow for selecting appropriate initial guess strategies based on molecular characteristics and convergence behavior:
Experimental Protocols for Challenging Cases
Protocol: Basis Set Projection for Large Basis Calculations
Purpose: To generate high-quality initial guesses for expensive calculations with large basis sets by leveraging pre-converged solutions in smaller basis sets.
Materials:
- Quantum chemistry software with basis set projection capabilities (e.g., Q-Chem with BASIS2 keyword) [16]
- Molecular structure coordinates
- Target level of theory and large basis set
- Appropriate smaller basis set for initial calculation
Procedure:
- Setup Initial Calculation: Configure a DFT calculation with the smaller basis set (BASIS2) using fast, stable functional (e.g., BP86)
- Converge Small Basis Calculation: Execute until full SCF convergence is achieved
- Project Density Matrix: Allow software to automatically project converged density matrix to larger target basis set
- Initialize Target Calculation: Begin main SCF calculation using projected guess
- Verify Convergence: Monitor SCF iterations for improved convergence behavior
Validation: Compare number of iterations required with and without projection; verify final energy matches expected accuracy benchmarks.
Protocol: Fragment-Based Initial Guess for Complex Systems
Purpose: To generate initial guesses for complex molecular systems by leveraging converged solutions from molecular fragments.
Materials:
- Quantum chemistry software with fragment MO capabilities (e.g., Q-Chem FRAGMO guess) [16]
- Structural data for complete system and logical fragments
- Pre-converged fragment calculations (if available)
Procedure:
- Fragment Identification: Decompose target molecule into logical fragments (functional groups, ligands, etc.)
- Fragment Calculation: Perform individual SCF calculations on each fragment
- Orbital Superposition: Use FRAGMO guess to superimpose converged fragment orbitals
- Occupancy Adjustment: Ensure proper orbital occupancy for combined system
- SCF Initiation: Begin target calculation using fragment-based guess
Validation: Check fragment calculations are properly converged; verify correct electron count in final system.
Research Reagent Solutions
Table 3: Computational Tools for Initial Guess Generation
| Tool/Software | Available Guess Methods | Specialized Capabilities | Typical Applications |
|---|---|---|---|
| Q-Chem | SAD, CORE, GWH, READ, FRAGMO, BASIS2 [16] | Basis set projection; fragment MO; orbital modification [16] | General purpose; large basis sets; complex molecules |
| ORCA | HCore, Hueckel, PAtom, PModel, MORead [17] | AutoStart feature; FMatrix/CMatrix projection; orbital rotation [17] | Transition metal complexes; open-shell systems |
| Psi4 | SAD, GWH, READ [5] | Integral tolerance adjustment; damping controls | Multireference systems; problematic geometries |
| Molpro | Various initial guesses | Advanced convergence algorithms | High-accuracy correlation methods |
Troubleshooting Pathological Convergence
Case Study: SCF Convergence at Pathological Geometries
A research case study highlights convergence challenges with a negatively charged molecule (H₃CClF) at pathological geometries characterized by widely separated atoms [5]. The SCF procedure showed oscillatory behavior where the energy appeared converged (ΔE < 1.14×10⁻¹³) but the density remained unconverged (RMS density ≈ 1.01×10⁻²) even after 200 iterations [5].
Successful mitigation strategies included:
- Integral Tolerance Adjustment: Setting
ints_tolerance 1.0E-16to reduce numerical noise [5] - Alternative Guess Methods: Using GWH guess instead of default SAD [5]
- Spin-State Modification: Converging higher spin state (septet) then projecting to target state [5]
- System Simplification: Attempting convergence in smaller basis sets before projection [5]
Advanced SCF Convergence Techniques
When improved initial guesses alone prove insufficient, the following advanced techniques can be combined with optimal guess selection:
- Damping and Level Shifting: Applying
SlowConvorVerySlowConvkeywords with percentage damping (e.g., 20%) or energy-based level shifting (e.g., 0.1) [1] - DIIS Optimization: Increasing
DIISMaxEqfrom default 5 to 15-40 for difficult cases to improve extrapolation [1] - Second-Order Convergence: Employing Trust Radius Augmented Hessian (TRAH) or Newton-Raphson (NR) algorithms when DIIS fails [1]
- Forced Fock Builds: Setting
directresetfreq 1to rebuild Fock matrix each iteration, eliminating numerical noise [1]
The selection of an appropriate initial guess is a critical determinant of SCF success, particularly for pathological systems including open-shell transition metal complexes, multireference systems, and molecules at unphysical geometries. Quantitative assessments demonstrate that Superposition of Atomic Potentials (SAP) generally provides the most robust initial guess, while specialized methods including basis set projection, fragment-based approaches, and orbital modification techniques offer powerful alternatives for challenging cases. By implementing the structured protocols and troubleshooting strategies outlined in this application note, researchers can significantly improve SCF convergence rates and computational efficiency in drug development and materials science applications.
Advanced SCF Algorithms and Software-Specific Implementations
Self-Consistent Field (SCF) convergence remains a fundamental challenge in electronic structure calculations, particularly for pathological systems such as open-shell transition metal complexes, metal clusters, and molecules with small HOMO-LUMO gaps [1] [18]. While traditional methods like Direct Inversion in the Iterative Subspace (DIIS) successfully converge most routine calculations, they frequently fail for these difficult cases, leading to oscillatory behavior or complete stagnation [1] [5]. This limitation has driven the development and implementation of more robust second-order convergence algorithms, including the Trust Region Augmented Hessian (TRAH), Newton-Raphson SCF (NRSCF), and Augmented Hessian SCF (AHSCF) methods.
Second-order convergence methods offer a fundamentally different approach from DIIS by utilizing both gradient and Hessian (second derivative) information to navigate the energy hyper-surface [19]. This provides superior convergence properties near the solution, typically achieving quadratic convergence rates compared to the linear or super-linear convergence of first-order methods [20]. For researchers investigating challenging chemical systems, understanding and appropriately deploying these advanced algorithms is crucial for obtaining physically meaningful results in a computationally efficient manner.
Theoretical Foundation of Second-Order Methods
The SCF Convergence Problem
The SCF procedure aims to find a set of molecular orbitals that minimize the total electronic energy under the constraint of orbital orthonormality. This optimization occurs in a space of orbital rotation parameters, which possesses a non-Euclidean, curved geometry [18]. The convergence difficulty arises from the complex topology of this energy landscape, which may contain multiple minima, saddle points, and regions of shallow curvature [19].
The key quantities in second-order convergence methods are:
- Orbital Gradient (G): The first derivative of the energy with respect to orbital rotations, which must vanish at convergence
- Orbital Hessian (H): The second derivative matrix that characterizes the curvature of the energy surface
- Error Vector: In DIIS, the commutator between the Fock and density matrices serves as an error vector [10]
For truly pathological systems, the energy landscape may contain regions where the Hessian has negative eigenvalues or near-zero eigenvalues, making navigation particularly challenging for simple optimization algorithms [19].
Fundamental Principles of Second-Order Algorithms
Second-order methods expand the energy as a Taylor series up to quadratic terms:
[ E(\kappa) \approx E(0) + \kappa^† G + \frac{1}{2} \kappa^† H \kappa ]
where (\kappa) represents the orbital rotation parameters. The minimum of this quadratic model provides the Newton step:
[ \kappa = -H^{-1} G ]
However, this pure Newton step is rarely used directly in SCF calculations due to the high computational cost of constructing and inverting the full Hessian, as well as its tendency to diverge when far from the solution [18]. Practical implementations employ sophisticated approximations and update strategies to make the approach computationally feasible while maintaining robustness.
Table 1: Comparison of SCF Convergence Algorithm Characteristics
| Method Type | Convergence Rate | Memory Requirements | Computational Cost per Iteration | Robustness for Pathological Cases |
|---|---|---|---|---|
| DIIS (First-Order) | Linear/Super-linear | Low to Moderate | Low | Poor |
| TRAH (Second-Order) | Quadratic | High | High | Excellent |
| NRSCF (Second-Order) | Quadratic | High | High | Good |
| AHSCF (Second-Order) | Quadratic | Moderate to High | Moderate to High | Good to Excellent |
Second-Order Convergence Algorithms
Trust Region Augmented Hessian (TRAH)
The Trust Region Augmented Hessian (TRAH) algorithm represents one of the most robust second-order convergers available in modern electronic structure codes like ORCA [1] [18]. TRAH combines the superior convergence properties of Newton-type methods with enhanced stability through a trust region approach that limits the step size to a region where the quadratic model is reliable.
The TRAH algorithm implements the following key features:
- Trust Region Control: Each step is constrained to lie within a "trust region" where the quadratic approximation remains valid
- Augmented Hessian Formulation: The orbital Hessian is expanded in a subspace of trial vectors, reducing computational cost
- Adaptive Convergence: The trust radius is automatically adjusted based on the accuracy of the quadratic model
In ORCA, TRAH is often activated automatically when the standard DIIS procedure encounters convergence difficulties [1]. This auto-TRAH feature can be fine-tuned through several parameters:
For particularly challenging cases, TRAH can be enforced from the beginning of the calculation using the ! TRAH keyword, bypassing the initial DIIS iterations entirely [18].
Newton-Raphson SCF (NRSCF) and Augmented Hessian SCF (AHSCF)
The Newton-Raphson SCF (NRSCF) method represents the direct application of Newton's method to the SCF problem, solving the exact Newton equations for the orbital rotation parameters [1]. While theoretically optimal in terms of convergence rate, the full NRSCF approach is computationally demanding due to the need to construct, store, and invert the complete orbital Hessian matrix.
The Augmented Hessian SCF (AHSCF) method addresses these limitations by:
- Working in a reduced subspace of the most important orbital rotations
- Diagonalizing an "augmented" matrix that contains both gradient and Hessian information
- Progressively expanding the subspace as convergence is approached
AHSCF typically offers an excellent balance between computational cost and convergence robustness, making it particularly suitable for large systems where the full Hessian would be prohibitively expensive [1].
Both NRSCF and AHSCF can be particularly effective for:
- Systems with near-degenerate orbitals
- Open-shell molecules with significant spin contamination
- Cases where DIIS exhibits oscillatory behavior
- Calculations requiring very tight convergence criteria
Relationship Between Algorithms
The following diagram illustrates the logical relationships and decision process for selecting between advanced SCF convergence algorithms:
Practical Implementation and Protocols
When to Use Second-Order Convergers
Second-order convergence methods are computationally more expensive per iteration than DIIS, but their superior convergence properties often lead to fewer overall iterations and greater reliability for challenging systems. The following scenarios warrant consideration of advanced convergers:
Transition Metal Complexes: Open-shell transition metal compounds represent one of the most common applications for second-order methods [1]. Their complex electronic structure with near-degenerate d-orbitals frequently causes DIIS failure. Both TRAH and NRSCF have demonstrated excellent performance for these systems.
Metal Clusters and Multi-Metallic Systems: Large clusters, particularly iron-sulfur clusters in bioinorganic chemistry, often require specialized SCF settings [1]. These systems may require combinations of second-order methods with damping and increased DIIS subspace size.
Radical Anions with Diffuse Functions: Conjugated systems with added electrons and diffuse basis functions present particular challenges due to near-linear dependencies in the basis set [1]. Second-order methods combined with full Fock matrix rebuilds can aid convergence.
Pathological Geometries: During geometry optimizations, particularly in the early stages, molecules may pass through regions of configuration space with near-degenerate orbitals or multiple competing electronic states [5]. These "pathological geometries" often benefit from second-order convergence algorithms.
Systems with Small HOMO-LUMO Gaps: Molecules with small band gaps, such as extended conjugated systems or donor-acceptor complexes, often exhibit convergence difficulties that respond well to second-order methods.
Implementation in ORCA: A Protocol
The following protocol outlines the systematic procedure for implementing second-order convergence methods in ORCA for pathological cases:
Protocol 1: TRAH Implementation for Open-Shell Transition Metal Complexes
Initial Assessment:
- Confirm DIIS failure through examination of the SCF output
- Check for oscillatory behavior in energy and density changes
- Verify that the molecular geometry is reasonable
Auto-TRAH Activation:
- Use default auto-TRAH settings initially:
! TightSCF - Monitor output for "TRAH activation" message
- Check convergence behavior after TRAH begins
- Use default auto-TRAH settings initially:
Manual TRAH Fine-Tuning (if needed):
- Implement custom TRAH parameters for problematic cases:
Convergence Verification:
- Confirm full convergence to
TolE < 1e-8(TightSCF standard) - Check spin contamination and other electronic properties
- Verify stability of the solution through SCF stability analysis
- Confirm full convergence to
Performance Optimization:
- For subsequent calculations, consider adjusting
AutoTRAHIterto balance DIIS and TRAH costs - If TRAH is too slow, consider using
! NoTRAHand implementing alternative strategies
- For subsequent calculations, consider adjusting
Protocol 2: NRSCF/AHSCF for Oscillatory Systems
Problem Identification:
- Recognize characteristic oscillatory pattern in SCF iterations
- Note large changes in orbital occupations or Fock matrix elements
Method Selection:
- For severe oscillations: implement
! NRSCF - For moderate oscillations with larger systems: use
! AHSCF - Combine with damping if oscillations persist:
! SlowConv
- For severe oscillations: implement
Parameter Optimization:
Convergence Monitoring:
- Watch for reduction in oscillation amplitude
- Verify monotonic convergence in later iterations
- Check final energy against expected chemical accuracy
Integration with Other SCF Enhancements
Second-order methods can be effectively combined with other SCF convergence techniques:
Initial Guess Improvement: For particularly difficult cases, converging a simpler method (e.g., BP86/def2-SVP) and reading the orbitals (! MORead) can provide a better starting point for second-order methods [1].
Damping and Level Shifting: Combining TRAH or NRSCF with damping (! SlowConv) or level shifting can help stabilize the early iterations [1].
DIIS Subspace Expansion: Before switching to full second-order methods, expanding the DIIS subspace can sometimes resolve convergence issues:
Performance Analysis and Optimization
Computational Considerations
The improved convergence robustness of second-order methods comes with increased computational costs that researchers must consider:
Memory Requirements: TRAH and NRSCF typically require 2-5 times more memory than standard DIIS due to storage of Hessian information and multiple trial vectors [18].
CPU Time per Iteration: Second-order iterations are significantly more expensive, with TRAH iterations typically 50-150% more computationally intensive than DIIS iterations.
Convergence Rate Trade-offs: Despite higher per-iteration costs, the reduced iteration count often makes second-order methods more efficient in wall time for difficult cases.
Table 2: Performance Characteristics of Second-Order Convergers
| Method | Typical Iteration Cost | Typical Iteration Count | Memory Overhead | Recommended System Size |
|---|---|---|---|---|
| DIIS | 1x (reference) | 20-50 | Low | All systems |
| TRAH | 1.5-2.5x | 10-30 | High | Small to medium |
| NRSCF | 2.0-3.0x | 5-20 | Very High | Small systems |
| AHSCF | 1.3-2.0x | 10-25 | Moderate | Medium to large |
Troubleshooting and Optimization
When second-order methods fail to converge or converge too slowly, consider these optimization strategies:
TRAH Parameter Adjustment:
- Reduce
AutoTRAHTOlto activate TRAH earlier - Increase
AutoTRAHNInterfor better interpolation - Adjust trust region parameters for better step control
Alternative Algorithm Selection:
- Switch from NRSCF to AHSCF for larger systems
- Consider KDIIS with SOSCF as an intermediate option [1]
- Implement geometric direct minimization (GDM) as an alternative [10]
System-Specific Settings: For truly pathological cases such as large metal clusters, combined approaches may be necessary [1]:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Advanced SCF Convergence
| Research Reagent | Function/Purpose | Implementation Example |
|---|---|---|
| TRAH Converger | Robust second-order convergence for pathological cases | ! TRAH in ORCA |
| NRSCF Algorithm | Exact Newton-Raphson implementation for quadratic convergence | ! NRSCF in ORCA |
| AHSCF Algorithm | Balanced approach with augmented Hessian methodology | ! AHSCF in ORCA |
| Auto-TRAH Parameters | Fine-tune automatic TRAH activation and performance | AutoTRAHTOl, AutoTRAHIter |
| SCF Convergence Criteria | Control precision of energy and density convergence | ! TightSCF, ! VeryTightSCF |
| DIIS Subspace Expansion | Enhance traditional DIIS for moderately difficult cases | DIISMaxEq 15-40 |
| Orbital Guess Options | Provide improved starting points for difficult convergences | ! PAtom, ! HCore, ! MORead |
| Damping Techniques | Stabilize initial SCF iterations | ! SlowConv, ! VerySlowConv |
| Level Shifting | Address convergence issues with near-degenerate orbitals | %scf Shift 0.1, ErrOff 0.1 end |
Second-order convergence methods represent essential tools in the computational chemist's arsenal for tackling challenging electronic structure problems. While DIIS remains the appropriate choice for most routine calculations, TRAH, NRSCF, and AHSCF provide robust alternatives for the pathological cases frequently encountered in research on transition metal complexes, open-shell systems, and molecules with complex electronic structures.
The key to successful implementation lies in understanding the specific convergence problems exhibited by a system and selecting the appropriate algorithm with optimized parameters. By systematically applying the protocols and strategies outlined in this application note, researchers can significantly enhance their ability to obtain converged SCF solutions for even the most challenging chemical systems.
As electronic structure theory continues to address increasingly complex chemical problems, the importance of robust convergence algorithms will only grow. Future developments in this field will likely focus on improving the computational efficiency of second-order methods while maintaining their convergence robustness, making them applicable to ever larger molecular systems.
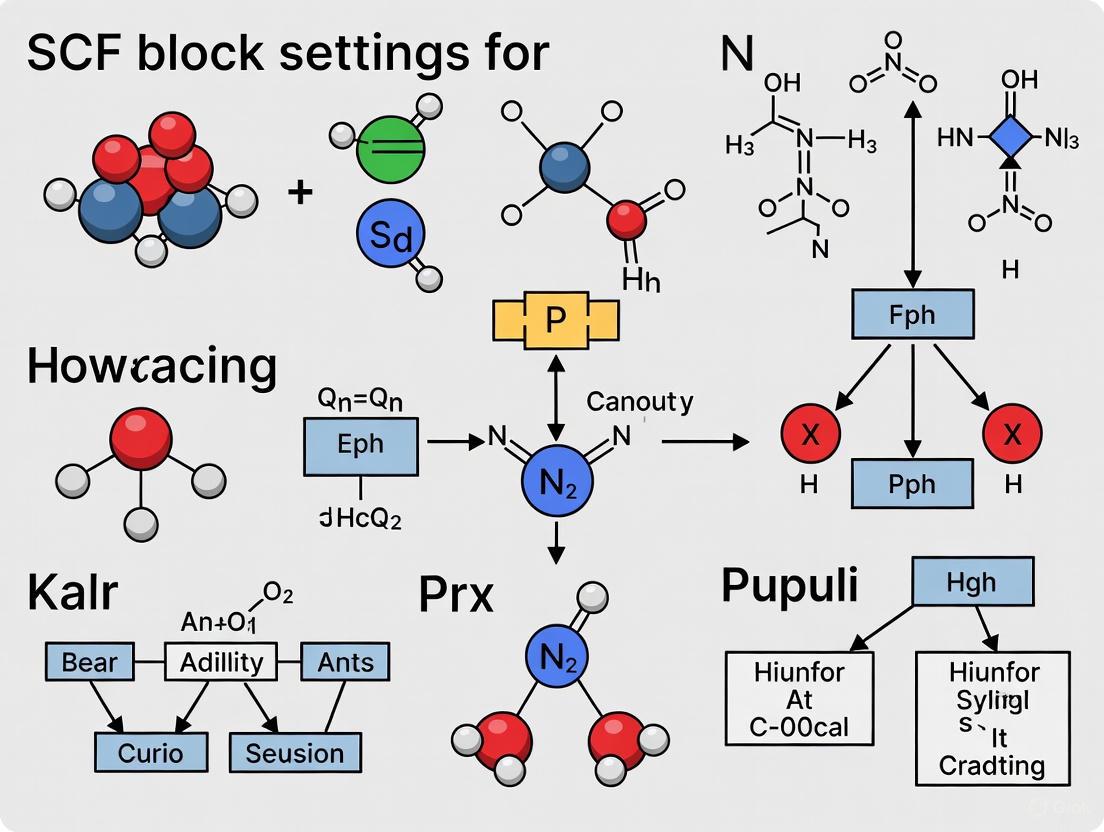
Self-Consistent Field (SCF) convergence represents a fundamental challenge in computational chemistry, particularly for pathological systems such as open-shell transition metal complexes, metal clusters, and conjugated radical anions with diffuse functions. Within the ORCA electronic structure package, such systems often defy standard convergence algorithms, necessitating specialized protocols and a deep understanding of advanced SCF settings. This application note, framed within a broader thesis on SCF block settings, provides a detailed guide for researchers and drug development professionals on employing the SlowConv, KDIIS, and TRAH parameters to overcome these pathological cases. We present structured quantitative data, step-by-step experimental protocols, and strategic workflows to guide users in selecting and tuning these critical parameters for robust SCF convergence.
Theoretical Background and Key Concepts
The SCF procedure iteratively solves the Hartree-Fock or Kohn-Sham equations until the electronic energy and density matrix stop changing significantly. Standard algorithms like DIIS (Direct Inversion in the Iterative Subspace) are highly efficient for well-behaved systems but can oscillate or diverge for pathological cases. ORCA 5.0 and later versions implement a sophisticated, automated SCF procedure that combines DIIS, the Second-Order SCF (SOSCF) method, and the Trust Radius Augmented Hessian (TRAH) approach. TRAH is a robust second-order converger that activates automatically when the primary DIIS-based procedure struggles, providing a more stable but computationally expensive path to convergence [1]. For cases where even this automation fails, understanding and manually configuring the SlowConv, KDIIS, and TRAH parameters becomes essential.
The Scientist's Toolkit: Essential SCF Parameters
Table 1: Key Research Reagent Solutions for Pathological SCF Convergence
| Item (Keyword/Block) | Primary Function | Typical Use Case |
|---|---|---|
! SlowConv / ! VerySlowConv |
Applies damping to control large energy/density oscillations in initial SCF cycles. | Transition metal complexes, open-shell systems with severe initial oscillations [1]. |
! KDIIS |
Uses the KDIIS algorithm as an alternative to standard DIIS for faster convergence. | Systems where standard DIIS trails or shows slow convergence; often used with SOSCF [1]. |
AutoTRAH Parameters |
Controls the automatic activation of the robust TRAH algorithm. | Systems where the default DIIS/SOSCF procedure fails to converge or struggles [1]. |
SOSCFStart |
Defines the orbital gradient threshold at which the SOSCF algorithm takes over. | Speeding up final convergence; crucial for some open-shell transition metal complexes [1]. |
DIISMaxEq |
Increases the number of previous Fock matrices used in DIIS extrapolation. | Pathological cases where DIIS struggles to find a good update direction (e.g., metal clusters) [1]. |
directresetfreq |
Controls how often the full Fock matrix is rebuilt to eliminate numerical noise. | Conjugated radical anions with diffuse functions or other numerically sensitive systems [1]. |
TightSCF |
Tightens SCF convergence tolerances (e.g., TolE 1e-8) for higher precision. |
Default in geometry optimizations; essential for reliable energies and properties [18] [21]. |
Quantitative Data and Parameter Tables
Precise control over numerical thresholds is critical. The following tables summarize key parameters for SCF convergence in ORCA.
Table 2: Standard SCF Convergence Tolerances (Selected) [18]
| Criterion | LooseSCF | NormalSCF | TightSCF | VeryTightSCF |
|---|---|---|---|---|
| TolE (Energy Change) | 1.0e-05 | 1.0e-06 | 1.0e-08 | 1.0e-09 |
| TolMaxP (Max Density Change) | 1.0e-03 | 1.0e-05 | 1.0e-07 | 1.0e-08 |
| TolRMSP (RMS Density Change) | 1.0e-04 | 1.0e-06 | 5.0e-09 | 1.0e-09 |
| TolG (Orbital Gradient) | 1.0e-04 | 5.0e-05 | 1.0e-05 | 2.0e-06 |
Table 3: Advanced SCF Parameters for Pathological Cases [1]
| Parameter | Default Value | Recommended for Pathological Cases | Effect on Calculation |
|---|---|---|---|
MaxIter |
125 | 500 - 1500 | Allows more iterations for slow convergence. |
DIISMaxEq |
5 | 15 - 40 | Improves DIIS extrapolation but increases memory use. |
directresetfreq |
15 | 1 - 5 | Reduces numerical noise; value of 1 is expensive. |
SOSCFStart |
0.0033 | 0.00033 | Triggers SOSCF algorithm earlier in the process. |
AutoTRAHTOl |
1.125 | Adjustable | Threshold for TRAH activation; lower for earlier activation. |
Experimental Protocols and Application Notes
Protocol 1: Employing SlowConv and Damping for Oscillatory Systems
Principle: The SlowConv and VerySlowConv keywords introduce damping to stabilize wild oscillations in the initial SCF cycles, which are common in metallic and open-shell systems [1].
Detailed Methodology:
- Initial Diagnosis: Monitor the SCF output. An oscillating or steadily increasing energy in the first 10-20 iterations indicates a need for damping.
- Basic Implementation: Add the keyword
! SlowConvto the input file. This applies a standard damping scheme. - Advanced Tuning with Level-Shifting: For persistent oscillations, combine
SlowConvwith a level-shift. - Execution and Verification: Run the calculation. Check the SCF output for a monotonic decrease in energy and orbital gradients. If convergence is still not achieved, proceed to
! VerySlowConvor integrate with the TRAH protocol (Section 4.3).
Protocol 2: Leveraging KDIIS and SOSCF for Accelerated Convergence
Principle: The KDIIS algorithm can sometimes achieve convergence faster than standard DIIS, especially when combined with the SOSCF method to refine the solution near convergence [1].
Detailed Methodology:
- Application: Use the keywords
! KDIIS SOSCFin the input file. - Handling SOSCF Instabilities: For open-shell systems or transition metal complexes, the SOSCF step can sometimes fail. If an error like "HUGE, UNRELIABLE STEP WAS ABOUT TO BE TAKEN" appears, delay the onset of SOSCF by reducing the
SOSCFStartthreshold. - Fallback Strategy: If instability persists, disable SOSCF with
! NOSOSCFand rely on KDIIS alone, or switch to the TRAH-based protocol.
Protocol 3: Configuring TRAH for Ultimate Robustness
Principle: TRAH is a second-order SCF converger that is more robust and reliable than DIIS-based methods but is also more computationally demanding. It can be manually forced or its auto-activation can be tuned [1].
Detailed Methodology:
- Manual Activation: To force the use of TRAH from the first iteration, use the keyword
! TRAH. - Tuning AutoTRAH: The default SCF procedure will automatically switch to TRAH if it detects problems. To fine-tune this behavior:
- Disabling TRAH: If TRAH is making the calculation unnecessarily slow and the system is converging well with DIIS, it can be disabled with
! NoTrah.
Protocol 4: Integrated Strategy for Truly Pathological Cases
Principle: For the most challenging systems, such as large iron-sulfur clusters, a combined approach using maximum damping, large DIIS space, and frequent Fock matrix rebuilds is necessary [1].
Detailed Methodology:
- Input Configuration:
- Alternative Guess Orbitals: If the above fails, generate an initial guess from a simpler, more robust method (e.g., BP86/def2-SVP) and read it in.
- Convergence Check: Verify that the
FINAL SINGLE POINT ENERGYline does not contain the warning "(SCF not fully converged!)" [1].
Workflow and Decision Diagrams
The following diagram outlines a logical diagnostic and treatment pathway for dealing with SCF convergence problems in ORCA, integrating the protocols detailed above.
Diagram 1: SCF Convergence Troubleshooting Workflow (76 chars)
Concluding Remarks and Best Practices
Successfully converging the SCF for pathological systems in ORCA requires a strategic, diagnostic approach. Begin with the simplest remedies, such as increasing MaxIter and using TightSCF, before moving to more specialized algorithms. The SlowConv keyword is your first line of defense against oscillations, while KDIIS can accelerate sluggish convergence. The TRAH algorithm represents the most robust tool in the arsenal and should be leveraged when other methods fail. Always remember to check the final energy message and geometry for convergence warnings. Combining these SCF strategies with a reasonable initial molecular geometry and, if needed, a better orbital guess (e.g., MORead) will maximize the likelihood of achieving a fully converged and physically meaningful result for even the most challenging chemical systems.
The Self-Consistent Field (SCF) method is a cornerstone of computational quantum chemistry, yet achieving convergence remains a significant challenge for many chemically interesting systems. Pathological convergence cases, such as open-shell transition metal complexes, systems with small HOMO-LUMO gaps, and molecules with strong static correlation, often defy standard convergence approaches like the default Pulay DIIS algorithm [10] [1]. Within the context of advanced research into SCF block settings for pathological cases, this application note details specialized algorithmic tools within Q-Chem's robust toolkit: Geometric Direct Minimization (GDM), Accelerated DIIS (ADIIS), and the Relaxed Constraint Algorithm (RCA) implemented in hybrid protocols [10] [22] [23]. These advanced algorithms and their strategic combination address different aspects of the SCF convergence problem, from poor initial guesses to oscillatory behavior near convergence, providing researchers with a systematic methodology for tackling even the most challenging systems encountered in computational drug development and materials science.
Table 1: Core SCF Algorithms for Pathological Convergence Cases in Q-Chem
| Algorithm | Primary Mechanism | Strengths | Ideal Use Cases |
|---|---|---|---|
| GDM | Geometric steps on hyperspherical orbital rotation space [24] [25] | High robustness, guaranteed convergence [24] | Restricted open-shell, DIIS failure cases [10] |
| ADIIS | Combination of ARH energy function and DIIS [23] | Fast initial convergence [26] | Poor initial guesses, early SCF cycles [26] |
| RCA | Relaxed idempotency constraint on density matrix [22] | Guaranteed energy decrease each cycle [22] | Core guess, symmetry breaking, near-degeneracy [22] |
| DIIS | Extrapolation from error vectors of previous cycles [10] | Fast convergence for well-behaved systems [10] | Standard default for most systems [10] |
Theoretical Foundations and Algorithmic Mechanisms
Geometric Direct Minimization (GDM): Navigating Orbital Space
The GDM algorithm fundamentally differs from DIIS-based approaches by directly minimizing the SCF energy with careful attention to the mathematical structure of the optimization space. GDM operates in an orbital rotation space that exhibits hyperspherical geometry—curved like a many-dimensional sphere [24] [25]. Traditional optimization methods that treat this space as flat Euclidean encounter efficiency and robustness limitations. GDM corrects this by taking steps along "great circles" (the optimal paths on spheres), analogous to how airplanes follow great circle routes rather than straight lines on flat maps [24]. This geometric propriety makes GDM extremely robust, though slightly less efficient than DIIS for well-behaved systems [24]. For restricted open-shell SCF calculations and systems where DIIS exhibits oscillatory behavior, GDM's mathematical foundation enables it to reliably converge where other methods fail [10] [25].
Relaxed Constraint Algorithm (RCA): Systematic Energy Descent
RCA addresses SCF convergence from a novel perspective by reformulating the constrained minimization problem. Standard SCF requires the density matrix to be idempotent (P·P = P), enforcing binary occupation numbers (0 or 1) [22]. RCA relaxes this constraint to allow sub-idempotent density matrices (P·P ≤ P), permitting fractional occupation numbers between 0 and 1 [22]. Mathematically, this relaxed constraint creates a convex optimization space where linear combinations of sub-idempotent matrices remain sub-idempotent. The algorithm minimizes the energy as a quadratic function of the density matrix, guaranteeing energy reduction at each iteration [22]. Physically, systems tend toward idempotency naturally as electrons fill lower-energy orbitals, making RCA particularly effective for poor initial guesses (e.g., core or GWH guesses) and cases with strong near-degeneracy effects where initial orbital occupancy is ambiguous [22].
Accelerated DIIS (ADIIS) and Hybrid Approaches
ADIIS combines the Augmented Roothaan-Hall (ARH) energy function with DIIS extrapolation to accelerate early-stage convergence [23]. While classical DIIS minimizes an error vector, ADIIS more directly targets energy lowering, making it effective when initial guesses are far from solution [26]. In practice, ADIIS excels in initial iterations but may become less efficient near convergence, making it ideal for hybrid approaches where it handles early convergence followed by a switch to more stable algorithms [26]. The fundamental strength of hybrid algorithms lies in combining complementary strengths: initial aggressive convergence followed by robust fine-tuning [24] [26] [22].
Experimental Protocols and Application Guidance
Protocol 1: DIIS_GDM for Final-Stage Convergence Failure
For systems where DIIS approaches the solution but fails to converge completely (oscillations near convergence), the DIIS_GDM hybrid algorithm is recommended [10] [23].
Step-by-Step Implementation:
- Algorithm Selection: Set
SCF_ALGORITHM = DIIS_GDMin the $rem section [10] [24] - Switching Threshold: The
THRESH_DIIS_SWITCHparameter controls when the algorithm switches from DIIS to GDM, defined as 10^-N [24] [25] - Iteration Limit: Set
MAX_DIIS_CYCLESto control the maximum number of DIIS iterations before switching [24] - Convergence Criteria: Set
SCF_CONVERGENCE = 8for geometry optimizations orSCF_CONVERGENCE = 5for single points [10] [23] - Integral Threshold: Ensure
THRESHis set at least 3 higher thanSCF_CONVERGENCE(e.g.,THRESH = 11withSCF_CONVERGENCE = 8) [10]
Example Implementation:
Mechanistic Rationale: This protocol leverages DIIS's efficiency at finding the general solution neighborhood, then employs GDM's robustness to finalize convergence, particularly effective for systems with challenging potential energy surface topology [24].
Protocol 2: RCA_DIIS for Poor Initial Guesses
When starting from poor initial guesses (core, GWH) or when symmetry breaking and near-degeneracy cause DIIS failure, RCA_DIIS provides superior performance [22] [23].
Step-by-Step Implementation:
- Algorithm Selection: Set
SCF_ALGORITHM = RCA_DIIS[10] [22] - Switching Control: Use
THRESH_RCA_SWITCHto define when to switch from RCA to DIIS (default 3, meaning switch at error < 10^-3) [22] - Iteration Limits: Set
MAX_RCA_CYCLESto control maximum RCA iterations [22] - Initial Guess: Employ
SCF_GUESS = GWHfor user-specified basis or problematic systems [22] - Special Considerations: Disable incremental Fock builds if convergence issues persist (
INCFOCK = FALSE) [22]
Example Implementation:
Mechanistic Rationale: RCA's guaranteed energy decrease at each iteration provides stable progress from poor initial guesses, while DIIS efficiently refines the nearly-converged solution [22].
Protocol 3: ADIIS_DIIS for Rapid Initial Progress
For systems where the initial guess is particularly poor and standard DIIS struggles to make progress, ADIIS_DIIS can provide rapid initial convergence [26] [23].
Step-by-Step Implementation:
- Algorithm Selection: Set
SCF_ALGORITHM = ADIIS_DIIS[23] - Switching Control: Use
THRESH_ADIIS_SWITCHandMAX_ADIIS_CYCLESto manage the transition [23] - Convergence Settings: Employ tighter thresholds for geometry optimizations (
SCF_CONVERGENCE = 8) [23]
Advanced Protocol: User-Customized Hybrid Algorithm
For maximum control over pathological cases, Q-Chem's GEN_SCFMAN enables sophisticated multi-algorithm workflows [26].
Step-by-Step Implementation:
- Enable Hybrid Algorithm: Set
GEN_SCFMAN_HYBRID_ALGO = TRUE[26] - Algorithm Sequencing: Define up to four algorithms and their transition criteria using
GEN_SCFMAN_ALGO_X,GEN_SCFMAN_ITER_X, andGEN_SCFMAN_CONV_X(where X = 1-4) [26] - Staged Convergence: Implement a sequence like ADIIS (early), DIIS (middle), GDM (final) for maximum robustness [26]
Example Implementation:
Table 2: Key Rem Variables for Advanced SCF Control
| Variable | Type | Default | Effect | Recommended Values |
|---|---|---|---|---|
| SCF_ALGORITHM | STRING | DIIS | Primary convergence method [10] | GDM, DIISGDM, RCADIIS [23] |
| DIISSUBSPACESIZE | INTEGER | 15 | Number of previous Fock matrices in DIIS [10] | 20-40 for difficult cases |
| THRESHDIISSWITCH | INTEGER | 2 | Switch threshold for DIIS_GDM (10^-N) [24] | 3-4 |
| MAXDIISCYCLES | INTEGER | 50 | Maximum DIIS cycles before switch [24] | 20-30 |
| THRESHRCASWITCH | INTEGER | 3 | Switch threshold for RCA_DIIS (10^-N) [22] | 3-4 |
| MAXRCACYCLES | INTEGER | 50 | Maximum RCA cycles before switch [22] | 20-40 |
| SCF_CONVERGENCE | INTEGER | 5/7/8 | Convergence threshold (10^-N) [23] | 8 (optimizations) [10] |
| GENSCFMANHYBRID_ALGO | BOOLEAN | FALSE | Enable multi-algorithm workflow [26] | TRUE for pathological cases |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Research Reagents for SCF Convergence
| Research Reagent | Function | Application Context |
|---|---|---|
| GWH Initial Guess | Generalized Wolfsberg-Helmholtz guess for molecular systems [22] | Poor initial guess situations, user-defined basis sets [22] |
| SAD Initial Guess | Superposition of Atomic Densities [24] | Standard initial guess compatible with DIIS_GDM [24] |
| Core Hamiltonian Guess | Diagonal Fock matrix elements [1] | Fallback option when other guesses fail |
| DIIS Subspace | History of previous Fock matrices for extrapolation [10] | Standard DIIS acceleration |
| Orbital Gradient | First derivative of energy with respect to orbital rotations [10] | Direct minimization methods (GDM, DM) |
| Approximate Hessian | Preconditioner for orbital optimization [27] | Second-order methods (Newton, GDM_LS) |
| Density Matrix | Central quantity in SCF procedure [10] | All SCF methods, particularly RCA |
Results and Discussion: Algorithm Performance in Pathological Cases
Comparative Analysis of Convergence Behavior
Each algorithm exhibits distinct convergence profiles suited to different stages of the SCF process and different types of pathological cases. DIIS demonstrates rapid initial convergence but may oscillate or diverge when near degeneracies or poor conditioning occur [10] [1]. GDM shows slower but more monotonic convergence, exceptionally reliable for final convergence stages [24] [25]. RCA provides guaranteed energy decrease each iteration, making it ideal for initial stages from poor guesses, though it may slow considerably near convergence [22]. ADIIS aggressively lowers energy initially but may become inefficient for final convergence [26]. The hybrid algorithms (DIISGDM, RCADIIS, ADIIS_DIIS) strategically combine these behaviors to overcome individual limitations [24] [22] [23].
Application to Specific Chemical Systems
Transition metal complexes represent particularly pathological cases due to dense orbital energy spectra and near-degeneracies [1]. For these systems, RCADIIS or ADIISDIIS protocols are recommended initially, as they effectively handle the poor starting guesses typical for d- and f-block elements [22] [1]. Open-shell systems with significant spin contamination benefit from GDM-based approaches, as the geometric optimization better handles the challenging orbital space [10] [25]. Extended conjugated systems with small HOMO-LUMO gaps often exhibit oscillatory convergence with DIIS but respond well to DIIS_GDM protocols that provide final convergence stability [24] [1].
Emerging Solutions: ROBUST Algorithm and Future Directions
Q-Chem 6.3 introduces the ROBUST and ROBUSTSTABLE algorithms as automated solutions for challenging convergence cases [23]. These black-box workflows implement tightened thresholds and strategic algorithm combinations (typically DIIS, ADIIS, and GDM) without requiring user intervention [23]. For drug development researchers dealing with diverse molecular systems, these automated approaches provide valuable fallback options when protocol-based solutions fail. The ROBUSTSTABLE variant additionally performs stability analysis to ensure the solution represents a true minimum rather than a saddle point [23]. As SCF methods continue evolving, the trend toward intelligent algorithm selection and hybridization represents the future of robust quantum chemical calculations for pathological systems.
Advanced SCF convergence algorithms in Q-Chem provide researchers with a powerful toolkit for addressing pathological cases that defy standard methods. Through strategic implementation of GDM, ADIIS, and RCA in hybrid protocols, even the most challenging systems—open-shell transition metals, near-degenerate systems, and complex excited states—can be systematically conquered. The protocols detailed herein establish a methodological foundation for SCF block settings specifically targeted at pathological convergence cases, enabling drug development researchers to advance their computational investigations without being impeded by SCF convergence failures. As quantum chemistry continues tackling increasingly complex chemical phenomena, these robust convergence strategies will remain essential tools in the computational scientist's arsenal.
Achieving self-consistent field (SCF) convergence is a fundamental challenge in computational electronic structure calculations, particularly for pathological systems such those with small HOMO-LUMO gaps, localized open-shell configurations (common in d- and f-element compounds), and transition state structures with dissociating bonds [13]. Within the ADF modeling suite, several advanced convergence accelerators have been implemented to address these difficult cases. This application note provides a detailed protocol for the selection and application of four key methods: MESA, LISTi, EDIIS, and the ARH method. The guidance is framed within broader research on SCF block settings aimed at resolving pathological convergence, with particular relevance for researchers investigating complex molecular systems in drug development and materials science.
The following table summarizes the core characteristics, operational mechanisms, and ideal use cases for the four primary convergence accelerators discussed in this note.
Table 1: Comparison of Key SCF Convergence Accelerators in ADF
| Method | Underlying Algorithm | Primary Strength | Typical Use Case | Performance Consideration |
|---|---|---|---|---|
| MESA | Multi-method hybrid (ADIIS, fDIIS, LISTb, LISTf, LISTi, SDIIS) [28] | High reliability through ensemble approach | Default choice for severely oscillating or stagnant convergence | Robust but requires evaluation of multiple algorithms |
| LISTi | Linear-expansion Shooting Technique [28] | Effectiveness for specific difficult electronic structures | Systems where default DIIS fails | Sensitivity to the number of DIIS expansion vectors (DIIS N); values of 12-20 can be effective [28] |
| EDIIS | Energy-DIIS (minimizes energy interpolation) [29] | Rapid initial progress from poor guess | Bringing a poor initial density guess into a convergent region | Combined with DIIS ("EDIIS+DIIS") for robustness [29] |
| ARH | Augmented Roothaan-Hall (direct energy minimization) [13] [29] | High reliability for minimization | Fallback option when other methods fail | Computationally more expensive; available with OldSCF [13] |
Method Selection and Implementation Workflow
The following diagram illustrates the decision pathway for selecting and applying the appropriate convergence accelerator based on the observed SCF behavior.
Detailed Experimental Protocols
Protocol 1: Implementing the MESA Method
The MESA (Multiple Acceleration Strategies Algorithm) method is a robust hybrid approach, recommended as a first step for pathological cases exhibiting oscillatory behavior.
Input Configuration for MESA:
Advanced Configuration: To deactivate a specific component within MESA (e.g., if SDIIS is suspected to cause instability), use:
Execution and Validation: Execute the calculation and monitor the SCF error in the output log. Successful convergence is indicated by a steady, monotonic decrease in the error. Persistent oscillation may require disabling specific MESA components.
Protocol 2: Configuring the LISTi Method
LISTi is a member of the Linear-expansion Shooting Technique family and can be effective where standard DIIS fails.
Input Configuration for LISTi:
Parameter Tuning: The DIIS N parameter controls the number of expansion vectors. For difficult systems, increasing this value to a range between 12 and 20 can be crucial for success [28]. However, for small molecules, an excessively large value can sometimes hinder convergence and should be tested.
Protocol 3: Employing EDIIS and ARH with OldSCF
The EDIIS and ARH methods are accessed through the OldSCF module, serving as a last resort for exceptionally stubborn cases.
Input Configuration for EDIIS+DIIS:
Input Configuration for ARH:
Theoretical Basis: The ARH method uses a quadratic augmented Roothaan–Hall energy function to obtain the linear coefficients for the density matrices in DIIS, directly minimizing the energy [29]. This differs from Pulay's DIIS, which minimizes the commutator of the Fock and density matrices.
Auxiliary Convergence Techniques
When the primary accelerators are insufficient, these auxiliary techniques can be combined to achieve stability.
Electron Smearing
This technique aids convergence in metallic systems or those with many near-degenerate levels around the Fermi energy by assigning fractional occupations.
Application Note: The Degenerate key introduces a smearing width (1e-4 Hartree in this example). To maintain physical results, the value should be kept as low as possible, and the calculation may be restarted with successively smaller values.
Stable DIIS Parameterization
For a slow but stable DIIS iteration, a conservative parameter set is recommended.
This configuration increases the number of DIIS vectors (N), delays the start of aggressive acceleration (Cyc), and reduces the mixing parameters to dampen the updates [13].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Computational Components for SCF Convergence
| Component | Function & Purpose | Implementation in ADF |
|---|---|---|
| SCF Block | Master input block regulating all SCF procedure aspects [28]. | SCF ... End |
| AccelerationMethod | Key subkey to select the core convergence algorithm [28]. | AccelerationMethod MESA |
| DIIS N | Controls the number of Fock/Density matrices in the iterative subspace [13] [28]. | DIIS N 15 End |
| Mixing / Mixing1 | Damping parameter for Fock matrix updates (first cycle / general) [13] [28]. | Mixing 0.015 |
| OldSCF | Keyword to enable the previous implementation of the SCF solver [28]. | OldSCF |
| Degenerate | Enables electron smearing to handle near-degenerate states [9]. | Convergence Degenerate 1e-4 End |
| Unrestricted | Switches to spin-unrestricted formalism for open-shell systems [30]. | Unrestricted Yes SpinPolarization 2.0 |
Application Notes
This document details specialized techniques for managing Self-Consistent Field (SCF) convergence in computationally challenging systems, such as open-shell transition metal complexes and materials exhibiting strong electron correlation. These methods are essential for researchers dealing with pathological convergence cases where standard SCF procedures fail.
Electron Smearing addresses convergence issues in metallic systems and small-gap semiconductors by artificially occupying bands around the Fermi level, preventing charge sloshing and oscillations in the SCF procedure. Level Shifting provides a more robust convergence pathway by artificially raising the energy of unoccupied orbitals. Forced Convergence techniques use rigorous algorithmic controls to push calculations to completion even when natural convergence stalls. Together, these methods form a critical toolkit for electronic structure calculations in drug development, particularly when modeling transition metal-containing enzymes or solid-state drug formulations where convergence pathologies are common.
Table 1: Standard SCF Convergence Tolerance Presets
The following compound keywords set multiple tolerance parameters simultaneously, providing a balanced approach for different precision needs [18].
| Convergence Preset | TolE (Energy) | TolRMSP (RMS Density) | TolMaxP (Max Density) | TolErr (DIIS Error) | Primary Application |
|---|---|---|---|---|---|
| LooseSCF | 1e-5 | 1e-4 | 1e-3 | 5e-4 | Preliminary geometry steps |
| NormalSCF | 1e-6 | 1e-6 | 1e-5 | 1e-5 | Standard single-point calculations |
| TightSCF | 1e-8 | 5e-9 | 1e-7 | 5e-7 | Transition metal complexes, property calculations |
| VeryTightSCF | 1e-9 | 1e-9 | 1e-8 | 1e-8 | High-precision spectroscopy, force constants |
Table 2: Smearing Methods and Parameters
Smearing techniques assign partial orbital occupations to stabilize the SCF cycle [31].
| Smearing Method | Functional Form | Width Parameter (σ) | Typical Use Case |
|---|---|---|---|
| Fermi-Dirac | Smooth, physical distribution | 0.01 - 0.10 eV | Metallic systems, accurate free energy |
| Gaussian | Gaussian broadening | 0.01 - 0.05 eV | General purpose density of states |
| Methfessel-Paxton | Order-N expansion | 0.05 - 0.20 eV | Rapid convergence in insulators/semiconductors |
Experimental Protocols
Protocol: Electron Smearing for Metallic Systems
This protocol is designed to achieve SCF convergence in systems with dense electronic states around the Fermi level, such as metals and narrow-gap semiconductors [31].
1. Initial Calculation Setup:
- Begin with a standard SCF calculation (
NormalSCF) without smearing to establish a baseline. - If oscillations in energy or density are observed over more than 20 cycles, proceed with smearing.
2. Selection and Application of Smearing:
- For most metallic systems, start with Fermi-Dirac smearing with a width of
σ = 0.01 eV(orkBT ≈ 0.00036 Ha). - In the ORCA input, specify the smearing type and width. Example for Fermi-Dirac:
- Perform the SCF calculation and monitor convergence.
3. Width Optimization and Final Calculation:
- If convergence is not achieved, incrementally increase the smearing width in steps of 0.01 eV up to a maximum of 0.10 eV.
- Critical Step: Once a converged geometry is obtained with smearing, a final single-point energy calculation must be performed without smearing or with a minimal smearing width (0.001 eV) to obtain the correct electronic energy for the ground state. The smeared energy is not the true ground-state energy.
Protocol: Level Shifting for Difficult Convergence
Level shifting is employed when the SCF cycle oscillates due to near-degeneracies between occupied and virtual orbitals [18].
1. Diagnosis of the Problem:
- Examine the SCF output for large orbital rotation angles or large orbital gradients. These indicate a poor starting point or near-degeneracies.
- Check for oscillations in the total energy that do not dampen over time.
2. Implementation of Level Shift:
- In the ORCA
%scfblock, activate level shifting. A typical initial shift value is 0.3 Hartree. - This technique works by artificially increasing the energy of the unoccupied orbitals, which dampens the orbital mixing that causes oscillations.
3. Progressive Reduction and Finalization:
- Run the SCF to convergence with the level shift active.
- For the final electronic energy, it is often necessary to gradually reduce the shift value to zero over several calculations to "guide" the system to the true minimum. Alternatively, use the shifted convergence as a stable starting point for an unshifted calculation.
Protocol: Forced Convergence with Tight Tolerances
This protocol uses stringent criteria and algorithmic forcing to handle the most pathological cases, such as open-shell transition metal complexes or broken-symmetry states [18].
1. Selection of Convergence Criteria:
- Use the
TightSCForVeryTightSCFkeyword to set rigorous tolerances for energy, density, and DIIS error. - Ensure the integral accuracy (
ThreshandTCut) is set tighter than the SCF density tolerance. A direct SCF cannot converge if the integral error is larger than the convergence criterion [18].
2. Configuration of the SCF Convergence Mode:
- In the
%scfblock, setConvCheckMode 0to require that all convergence criteria are met. This is the most rigorous setting. - Activate
ConvForced 1to prevent the calculation from proceeding to the next step (e.g., a geometry optimization) without first achieving SCF convergence.
3. Advanced Algorithm Selection:
- For extremely difficult cases, switch to the Trust-Region Augmented Hessian (TRAH) SCF solver by using the
!TRAHkeyword. This algorithm is more robust and is guaranteed to find a local minimum.
Visualization of Workflows
Diagram 1: SCF Convergence Technique Decision Pathway
This diagram outlines the logical process for selecting and applying the appropriate convergence technique based on SCF behavior.
Diagram 2: Protocol for Electron Smearing and Energy Correction
This workflow details the specific steps for applying the electron smearing protocol, including the critical final energy correction step.
Research Reagent Solutions
Table 3: Computational Toolkit for Pathological SCF Cases
This table lists key software, algorithms, and parameters that function as essential "research reagents" for resolving SCF convergence issues.
| Reagent / Solution | Function / Purpose | Example Usage & Notes |
|---|---|---|
| Fermi-Dirac Smearing | Smears electronic occupation to stabilize metallic systems. | ! Fermi-Dirac 0.01 in ORCA. Prevents charge sloshing [31]. |
| Methfessel-Paxton Smearing | Approximate smearing for faster convergence in insulators. | ! Methfessel-Paxton 0.05. Can be less accurate for metals [31]. |
| Level Shift Parameter | Artificially raises virtual orbital energies. | %scf Shift ShiftValue 0.3 end. Dampens orbital mixing [18]. |
| TRAH SCF Solver | Robust, guaranteed-convergence algorithm. | !TRAH keyword. Replaces default DIIS in extreme cases [18]. |
| DFT+U Correction | Adds electron correlation for localized d/f electrons. | Essential for transition metal complexes in spin crossover studies [32] [33]. |
| Forced Convergence (ConvForced) | Halts calculation if SCF fails. | %scf ConvForced 1 end. Prevents use of unconverged wavefunctions [18]. |
A Systematic Troubleshooting Protocol for Stubborn SCF Cases
The Self-Consistent Field (SCF) procedure is an iterative computational method fundamental to Kohn-Sham density functional theory, seeking a solution where the electronic density remains consistent with the potential it generates [9]. For researchers in drug development investigating complex molecular systems such as transition metal complexes or open-shell species, achieving SCF convergence is a prerequisite for obtaining reliable energy, structure, and property predictions. Pathological cases, where standard convergence approaches fail, necessitate a rigorous and systematic diagnostic protocol. This Application Note details a step-by-step framework, from initial geometry validation to advanced wavefunction and gradient analysis, to diagnose and correct SCF convergence failures, framed within broader research on SCF block settings for pathological cases.
Initial Diagnostic Steps: Geometry and Guess
Geometry Integrity Check
The diagnostic protocol begins with an assessment of the initial molecular geometry, as an unreasonable structure is a primary cause of SCF failure [1].
- Bond Lengths and Angles: Verify that all bond lengths and angles are chemically plausible. Particular attention should be paid to transition metal-ligand distances.
- Molecular Symmetry: High symmetry can sometimes lead to orbital degeneracy and convergence challenges. A slight distortion of the geometry (e.g., by 0.01 Å) can break symmetry and aid convergence.
- Steric Clashes: Identify any unrealistically short non-bonded contacts that could create an unphysical initial electron density.
Initial Guess and Wavefunction Stability
The starting guess for the molecular orbitals critically influences the SCF trajectory [1].
- Alternative Guess Strategies: If the default PModel guess fails, consider more robust alternatives such as the
PAtom(potential atom superposition),Hueckel, orHCoreguesses. - Converging a Simpler System: A highly effective strategy is to first converge the SCF for a simpler model system or method (e.g., BP86/def2-SVP or HF/def2-SVP). The resulting orbitals can then be used as a guess for the target calculation via the
MOReadkeyword [1]. - Oxidized/Reduced State Convergence: For open-shell systems, attempt to converge a closed-shell, 1- or 2-electron oxidized or reduced state. The orbitals from this solution can provide a superior starting point for the target electronic state [1].
Table 1: Initial Guess Strategies for Pathological Systems
| Strategy | Key Input/Keyword | Primary Use Case | Rationale |
|---|---|---|---|
| Simpler Method/Basis | ! BP86 def2-SVP |
All pathological systems | Provides a stable, initial wavefunction from a less expensive, more robust calculation [1]. |
| Orbital Reading | ! MORead %moinp "guess.gbw" end |
Systems with known stable wavefunctions from a previous calculation | Uses a pre-converged set of orbitals, bypassing the initial guess problem. |
| Core Hamiltonian Guess | %scf Guess HCore end |
Systems where default guess fails | Generates initial orbitals from a calculation ignoring electron-electron repulsion. |
| Converged Ion Guess | Manually create ion input file | Open-shell systems struggling to find a stable state | A closed-shell ion often converges more readily, providing a good orbital starting point [1]. |
SCF Convergence Parameter Tuning
Convergence Criteria and Iteration Limits
The default SCF convergence criterion and maximum iteration limit may be insufficient for difficult cases [9] [1].
- Tightening Criteria: For highly accurate single-point energies, use a
TightSCFkeyword or equivalent to enforce a more stringent convergence threshold. - Increasing Iterations: The first response to slow but progressive convergence should be to increase the maximum number of SCF cycles significantly (e.g.,
%scf MaxIter 500 end) [1]. - Modest Criterion: In geometry optimizations, if the SCF is consistently near convergence but not fully converged, using a
ModestCriterionallows the optimization to proceed, often resolving itself in later cycles [9].
Advanced SCF Algorithms and Damping
When basic damping fails, advanced algorithms offer a path to convergence.
- Second-Order Convergers: The Trust Radius Augmented Hessian (TRAH) or Newton-Raphson (NRSCF) algorithms are robust but expensive second-order methods that can handle cases where first-order methods fail [1]. TRAH may activate automatically in modern codes like ORCA upon detecting SCF oscillations.
- KDIIS with SOSCF: The KDIIS (Kramers-augmented Direct Inversion in the Iterative Subspace) algorithm, sometimes combined with the Superposition-of-Configurations (SOSCF) method, can enable faster convergence for some transition metal complexes [1].
- Damping and Level Shifting: For wild oscillations in the initial SCF iterations, increased damping (
! SlowConvor! VerySlowConv) is essential. Level shifting (e.g.,%scf Shift Shift 0.1 ErrOff 0.1 end) can stabilize convergence by shifting unoccupied orbitals to higher energy, reducing near-degeneracy issues [1].
Table 2: SCF Algorithm Selection Guide for Pathological Cases
| Algorithm/Setting | Key Input/Keyword | Strengths | Computational Cost |
|---|---|---|---|
| Standard DIIS | Default in many codes | Fast, efficient for well-behaved systems | Low |
| TRAH | ! TRAH or auto-activated |
Highly robust, handles severe oscillations and near-degeneracies | High |
| KDIIS + SOSCF | ! KDIIS SOSCF |
Effective for many transition metal complexes [1] | Medium |
| Damping (SlowConv) | ! SlowConv |
Suppresses large oscillations in early iterations [1] | Low (but may slow convergence) |
| Level Shifting | %scf Shift Shift 0.1 end |
Stabilizes convergence by addressing near-degeneracies | Low |
The following workflow diagram outlines the logical decision process for diagnosing and treating SCF convergence issues.
SCF Diagnosis Workflow
Wavefunction Analysis and Gradient Diagnostics
Orbital and Gradient Analysis
When algorithmic changes are insufficient, direct analysis of the wavefunction and its gradients is required.
- Orbital Gradient Monitoring: Monitor the maximum (MaxP) and root-mean-square (RMSP) elements of the orbital gradient. A trailing convergence where the energy change (
DeltaE) is small but gradients are large indicates a shallow energy surface or near-degeneracy [1]. - DIIS Space Management: In cases of severe oscillation, increasing the size of the DIIS subspace (
DIISMaxEq 15) provides a longer memory for extrapolation, which can stabilize convergence. For numerically noisy systems, increasing the frequency of Fock matrix rebuilds (directresetfreq 1) can help [1]. - Electronic Smearing: Using the
Degeneratekey or applying a finite electronic temperature (ElectronicTemperature) smears orbital occupations around the Fermi level. This is particularly effective for metallic systems or those with (near-)degenerate HOMO-LUMO gaps, as it smooths the energy landscape [9].
Gradient Nonlinearity and Conditioning
In extreme cases, the fundamental mathematical problem becomes ill-conditioned.
- Elongated Systems: Systems with very non-cubic cell dimensions (e.g., slabs, nanotubes) ill-condition the charge-mixing problem. This often requires a significant reduction of the mixing parameter (
MixingorAMIX) [4]. - Spin Density Mixing: For antiferromagnetic or noncollinear magnetic systems, convergence is notoriously difficult. Simultaneously reducing the charge (
AMIX) and spin density (AMIX_MAG) mixing parameters to low values (e.g., 0.01) is often necessary [4].
Table 3: Troubleshooting Guide Based on SCF Symptomology
| SCF Symptom | Probable Cause | Corrective Actions | Key Parameters to Adjust |
|---|---|---|---|
| Wild oscillations from cycle 1 | Poor initial guess, strong near-degeneracies | Strong damping, level shifting, improved guess | !SlowConv, Shift, Guess |
| Convergence is slow but steady | Insufficient iterations, slow convergence rate | Increase iterations, use convergence accelerators | MaxIter, !SOSCF |
| Convergence trails (small DeltaE, large gradients) | Shallow energy minimum, numerical noise | SOSCF, Fock matrix rebuild, smearing | SOSCFStart, directresetfreq, Degenerate |
| Convergence stalls at high error | Wrong electron state, symmetry issues | Change spin state, break symmetry, use MORead | SpinFlip, MORead |
The Scientist's Toolkit: Research Reagent Solutions
This table catalogs essential computational "reagents" and their functions for resolving SCF convergence pathologies.
Table 4: Essential Research Reagents for SCF Convergence
| Reagent / Keyword | Function / Purpose | Application Context |
|---|---|---|
!SlowConv / !VerySlowConv |
Applies increased damping to suppress large density oscillations in early SCF cycles [1]. | Systems with initial wild oscillations (common in open-shell TM complexes). |
!TRAH |
Activates the Trust Radius Augmented Hessian, a robust second-order SCF converger [1]. | Default DIIS failure; systems with severe convergence pathologies. |
!KDIIS SOSCF |
Uses Kramers DIIS with Superposition-of-Configurations to accelerate convergence [1]. | Alternative for transition metal complexes where standard DIIS fails. |
Degenerate / ElectronicTemperature |
Smears orbital occupations near the Fermi level, smoothing the energy landscape [9]. | Metals, systems with small HOMO-LUMO gaps, or near-degeneracies. |
MORead |
Reads initial molecular orbitals from a previous calculation, bypassing the initial guess problem [1]. | Restarting calculations or using orbitals from a simpler, pre-converged system. |
DIISMaxEq |
Increases the number of previous Fock matrices used in DIIS extrapolation, improving stability [1]. | Cases where DIIS extrapolation becomes unstable (e.g., oscillations). |
directresetfreq |
Controls how often the exact Fock matrix is rebuilt, reducing numerical noise [1]. | Calculations where numerical integration inaccuracies hinder convergence. |
Self-Consistent Field (SCF) convergence remains a fundamental challenge in electronic structure theory, particularly for pathological systems such as open-shell transition metal complexes, metal clusters, and conjugated radical anions. These systems often exhibit strong electronic degeneracies, small HOMO-LUMO gaps, and pronounced spin polarization, which can lead to oscillatory behavior or complete divergence in standard SCF algorithms. Within the broader context of developing robust SCF protocols for difficult cases, this application note provides a detailed examination of three critical parameters: DIIS subspace size, damping factors, and direct reset frequency. Based on comprehensive research across multiple computational chemistry packages, we present structured quantitative data and experimental protocols to guide researchers in systematically tuning these parameters to achieve convergence in otherwise intractable systems.
Theoretical Background and Key Parameters
The SCF procedure seeks a convergent solution to the Roothaan equations, F C = S C E, through an iterative process. For well-behaved systems, default algorithms like Pulay's DIIS (Direct Inversion in the Iterative Subspace) provide rapid convergence. However, pathological cases require careful parameter adjustment to stabilize the convergence pathway.
The DIIS Mechanism and Subspace Management
The DIIS method extrapolates a new Fock matrix by forming a linear combination of previous Fock matrices, Fk = Σ cj Fj, where the coefficients cj are determined by constrained minimization of the DIIS error vector, typically the commutator [F, PS] [34] [10]. The size of the DIIS subspace—the number of previous Fock matrices stored for extrapolation—is a critical parameter. While a larger subspace can capture more complex convergence trajectories, it also increases memory usage and can become ill-conditioned near convergence.
Damping and Direct Reset Dynamics
Damping is a simpler, older technique that stabilizes the SCF process by linearly mixing the current density matrix with that from the previous iteration: Pdamped = (1-α)Pn + αP_(n-1) [35]. This reduces large oscillations in the early iterations. The direct reset frequency controls how often the full Fock matrix is rebuilt from scratch, eliminating numerical noise that can accumulate in direct SCF methods and hinder convergence [1].
Quantitative Parameter Tables
Table 1: DIIS Subspace Size Recommendations for Various System Types
| System Type | Recommended DIIS Subspace Size | Default in Typical Software | Rationale |
|---|---|---|---|
| Standard Organic Molecules (Closed-Shell) | 5-10 [1] | 15 (Q-Chem) [34] | Sufficient for typical convergence; balances speed and stability |
| Pathological Cases (TM Complexes, Clusters) | 15-40 [1] | 15 (Q-Chem) [34] | Larger history provides better extrapolation in oscillatory systems |
| Extremely Difficult Cases (e.g., Fe-S Clusters) | Up to 40 [1] | N/A | Maximum stabilization for systems with severe convergence issues |
Table 2: Damping and Direct Reset Frequency Parameters
| Parameter | Typical Default | Recommended for Problematic Cases | Effect |
|---|---|---|---|
| Damping Factor (α) | Not applied by default [35] | 0.5 - 0.75 (NDAMP = 50-75) [35] | Reduces large density matrix fluctuations in early cycles |
| Damping Duration | 3 cycles (Q-Chem) [35] | 20+ cycles or until fluctuation subsides [1] [35] | Provides sustained stabilization in highly oscillatory systems |
| Direct Reset Frequency | 15 (ORCA) [1] | 1 (most expensive) or 1-15 [1] | Rebuilds Fock matrix, eliminating numerical noise that impedes convergence |
Table 3: Comprehensive SCF Convergence Tolerances (ORCA)
| Criterion | !LooseSCF | !NormalSCF | !TightSCF | !VeryTightSCF |
|---|---|---|---|---|
| TolE (Energy Change) | 1e-5 | 1e-6 | 1e-8 | 1e-9 |
| TolMaxP (Max Density Change) | 1e-3 | 1e-5 | 1e-7 | 1e-8 |
| TolRMSP (RMS Density Change) | 1e-4 | 1e-6 | 5e-9 | 1e-9 |
| TolErr (DIIS Error) | 5e-4 | 1e-5 | 5e-7 | 1e-8 |
| Recommended Use Case | Preliminary scans | Standard single-point | Geometry optimizations, frequencies | High-precision property calculations |
Experimental Protocols
Protocol 1: Standard Approach for Moderately Difficult Cases
This protocol is suitable for systems where the default DIIS procedure shows oscillatory behavior but does not diverge catastrophically.
- Initial Setup and Diagnosis: Run an SCF calculation with default settings. Monitor the convergence profile. If consistent, slow oscillations in energy or density are observed, proceed with this protocol.
- Increase DIIS Subspace: Set the DIIS subspace size to 20-25. This allows the algorithm to utilize a longer history for extrapolation, which can break cyclic convergence patterns [1].
- Apply Moderate Damping: Introduce damping for the initial 10-15 cycles with a mixing factor α = 0.5 (NDAMP = 50 in Q-Chem) [35]. This stabilizes the early iterations without significantly slowing down later convergence.
- Switch to Standard Algorithm: After the initial damped cycles, allow the standard DIIS or a second-order algorithm to take over for fine convergence.
Diagram 1: Protocol for Moderately Difficult Cases
Protocol 2: Advanced Protocol for Pathological Systems
For highly pathological systems (e.g., open-shell transition metal clusters, conjugated radical anions with diffuse functions), a more aggressive strategy is required.
- Initial Guess Enhancement: Begin with a high-quality initial guess. Converge a simpler, closed-shell system (e.g., a different charge state) or use a low-level method (e.g., BP86/def2-SVP) and read the resulting orbitals in for the target calculation using the
MOReadkeyword in ORCA orinit_guess = chkin PySCF [11] [1]. - Aggressive DIIS and Damping: Combine a large DIIS subspace (30-40) with strong damping (α = 0.75, NDAMP = 75). Maintain damping for a longer duration (e.g., 20 cycles or until the energy change falls below a threshold of 1e-3) [1] [35].
- High-Frequency Direct Reset: Set the direct reset frequency to 1, forcing a full rebuild of the Fock matrix in every iteration. This is computationally expensive but eliminates numerical noise that is often the hidden cause of non-convergence in direct SCF methods [1].
- Fallback to Second-Order Optimizer: If the above steps fail, abandon the DIIS-based approach and switch to a robust second-order convergence algorithm, such as the Trust Region Augmented Hessian (TRAH) in ORCA [1] [18] or the geometric direct minimization (GDM) in Q-Chem [10]. These methods are more robust but have a higher computational cost per iteration.
Diagram 2: Protocol for Pathological Systems
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Software Tools and Algorithms for SCF Convergence Research
| Tool / Algorithm | Function in Convergence Research | Software Availability |
|---|---|---|
| DIIS (Pulay) | Primary extrapolation driver; accelerates convergence by minimizing the error vector from previous cycles [34]. | Q-Chem, PySCF, ORCA |
| GDM/TRAM | Robust fallback optimizers; use geometric direct minimization or trust region methods to ensure convergence when DIIS fails [10] [1] [36]. | Q-Chem, ORCA, OpenTrustRegion |
| Damping Algorithm | Stabilizing agent; suppresses large oscillations in the early SCF iterations by mixing old and new density matrices [35]. | Q-Chem, ORCA |
| Stability Analysis | Diagnostic tool; checks if a converged wavefunction is a true minimum or an unstable saddle point [11]. | PySCF, ORCA |
| Linear Prediction Extrapolators | Advanced guess generator; uses signal processing techniques on previous MD timesteps to create a superior initial Fock matrix guess [37]. | Custom implementations |
Achieving SCF convergence for pathological systems is a common hurdle in computational drug development and materials science. A deep understanding and systematic tuning of DIIS subspace size, damping factors, and direct reset frequency can resolve the majority of these challenges. The quantitative data and step-by-step protocols provided here offer a structured methodology for researchers to diagnose and treat SCF convergence failures. The recommended approach is progressive: start with moderate adjustments to the standard DIIS algorithm and escalate to more aggressive, resource-intensive protocols only when necessary, ultimately falling back to robust second-order optimizers for the most stubborn cases. This structured tuning of SCF block settings ensures reliability and expands the scope of computable molecular systems.
Iron-sulfur (Fe-S) clusters and synthetic transition metal complexes represent two major classes of metalloeffectors with significant biological and therapeutic implications. Fe-S clusters are ancient, ubiquitous protein cofactors that drive essential cellular functions from electron transport to enzyme catalysis, but their intrinsic sensitivity to oxidation renders them vulnerable in various pathological conditions [38]. Simultaneously, transition metal complexes have emerged as strategic therapeutic candidates to combat the global crisis of antimicrobial resistance and other diseases, offering multifactorial mechanisms for microbial disruption that can overcome conventional drug resistance [39]. The intersection of these systems creates a unique pathological landscape where synthetic metal complexes can target Fe-S cluster-dependent pathways in pathogens while Fe-S clusters themselves function as crucial sensors in human disease states.
The redox sensitivity of Fe-S clusters has shaped the evolution of specialized biosynthetic and protective mechanisms [38]. Recent findings highlight how human Fe-S-binding regulators exploit this cofactor's reactivity to sense iron and oxygen levels, translating environmental cues into appropriate homeostatic responses [38]. Understanding these intersecting systems provides critical insights for developing novel therapeutic strategies against evolving pathogens and metalloenzyme-related disorders.
Table: Key Characteristics of Metalloeffectors in Pathological Systems
| Feature | Iron-Sulfur Clusters | Transition Metal Complexes |
|---|---|---|
| Primary Role | Biological cofactors in electron transfer, catalytic reactions | Therapeutic agents against resistant pathogens, cancer |
| Pathological Significance | Vulnerability to oxidative stress; sensors in disease states | Overcoming drug resistance through multiple mechanisms |
| Key Vulnerabilities | Oxygen sensitivity; disassembly under oxidative conditions | Potential toxicity; metabolic complications |
| Therapeutic Targeting | Antibacterial drug targets (e.g., sulfonucleotide reductases) | Direct antimicrobial, anticancer applications |
Iron-Sulfur Clusters: Biological Roles and Pathological Significance
Physiological Functions and Sensing Mechanisms
Iron-sulfur clusters are critical to a wide range of biological processes, from DNA repair and transcriptional regulation to mitochondrial respiration and enzymatic catalysis [40]. Their unique ability to facilitate electron transfer, catalyze reactions involving organic radicals, and stabilize protein structures makes them indispensable across all domains of life [40]. Composed of iron and inorganic sulfur, the most common Fe-S cluster types are rhombic [2Fe-2S] and cubane [4Fe-4S] structures, each with distinct redox properties and functional capabilities [40].
Well-tuned to cellular iron and oxygen status, Fe-S clusters serve as ideal environmental sensors [38]. Mammalian cells contain at least four known iron and oxygen-response proteins dependent on Fe-S cluster sensors: iron response protein 1 (IRP1), the E3 ubiquitin ligase FBXL5, nuclear receptor coactivator 4 (NCOA4), and the outer mitochondrial membrane protein CISD1 (mitoNEET) [38]. These sensors translate environmental cues into homeostatic responses through cluster-dependent conformational changes. For instance, IRP1 functions as cytosolic aconitase when bound to a [4Fe-4S] cluster but transforms into a translational regulator in its cluster-free state, modulating transcripts involved in iron homeostasis [38].
Pathological Disruptions and Disease Connections
The redox sensitivity of Fe-S clusters renders them particularly vulnerable to pathological conditions characterized by oxidative stress. Multiple disorders disrupt normal tissue oxygen thresholds, including ischemia-reperfusion injury, obstructive sleep apnea, bronchopulmonary dysplasia, and mitochondrial diseases [38]. Recent research has delineated four human pathways especially vulnerable to high oxygen tensions: purine metabolism, diphthamine synthesis, nucleotide excision repair, and electron transport chain activity [38]. Each of these pathways contains highly labile Fe-S cluster proteins that become impaired under hyperoxic conditions.
An increasing number of human conditions are being identified through exome sequencing that are caused by loss of function in components of the Fe-S biogenesis machinery [40]. Pathogenic variants in Fe-S domains of DNA helicases like XPD, FANCJ, RTEL1, DDX11, and glycosylases such as NTHL1 and MUTYH have been linked to several cancers and compromised DNA repair activity [40]. Furthermore, the relevance of radical S-adenosyl-L-methionine (RS) enzymes containing [4Fe-4S] clusters to human health is paramount, with gene variants linked to diseases like molybdenum cofactor deficiency, lipoic acid deficiencies, type 2 diabetes, and motor neuron degeneration in ALS [40].
Fe-S Cluster Disruption in Human Disease: This diagram illustrates how oxidative stress, hyperoxia, and genetic mutations disrupt Fe-S cluster integrity, leading to pathway dysfunction and disease.
Transition Metal Complexes as Therapeutic Agents
Antimicrobial Applications and Resistance Mechanisms
Transition metal complexes are gaining prominence as strategic antimicrobial candidates to combat the global crisis of microbial resistance, driven by the declining efficacy of conventional antibiotics [39]. Their attributes, originally leveraged for catalysis, photoelectric applications, and biocompatible designs, now enable multifactorial mechanisms for microbial disruption. These properties allow cationic species to destabilize bacterial membranes, redox-active surfaces to generate bactericidal reactive oxygen species (ROS), and programmable coordination architectures to target pathogen-specific vulnerabilities [39].
The bioactivity profiles of metal complexes are intrinsically linked to their electronic structures. Redox-active metals (e.g., Fe, Cu, Co) frequently display high cytotoxicity due to Fenton-type reactions that generate indiscriminate ROS in mammalian cells, while d8/d6 low-spin metals (e.g., Ru(II/III), Ir(III), Pt(II)) demonstrate higher selectivity due to their kinetic inertness and tunable ligand fields that minimize off-target interactions [39]. This electronic-toxicity correlation underscores that optimizing metal-centered properties (oxidation state, d-electron count, ligand field strength) is critical for enhancing therapeutic indices.
Table: Antimicrobial Transition Metal Complexes and Their Applications
| Metal Complex | Target Pathogens | Key Mechanisms | Advantages |
|---|---|---|---|
| Silver Sulfadiazine | Gram-positive (S. aureus) and Gram-negative (P. aeruginosa) bacteria | DNA binding; multi-target effects; controlled Ag+ release | Broad-spectrum activity; topical application for burn wounds |
| Copper Complexes | Drug-resistant bacteria; fungal pathogens | Redox properties; affinity for biological ligands | Nonspecific targeting; multiple disruption mechanisms |
| Ruthenium Complexes | Cancer cells; microbial infections | Covalent binding to biomolecules; enzyme inhibition | Tunable ligand fields; selective toxicity |
| Gold Complexes (Auranofin) | Rheumatoid arthritis; parasitic infections | Binding to cysteine/selenocysteine residues in enzymes | Repurposing potential; multiple therapeutic applications |
Molecular Mechanisms of Action
Transition metal complexes employ three primary mechanisms of action: covalent binding to biomolecules, inhibition of enzymes, and redox activity [41]. Due to their unique capability to exchange ligands and stabilize different geometries, metal-based drugs are uniquely equipped for covalent binding to biomolecules, as exemplified by cisplatin and its analogues which coordinate to N(7) of guanine in DNA [41]. Alternatively, some metal complexes preferentially bind to enzymes rather than DNA, with gold-based compounds like auranofin covalently binding to cysteine and selenocysteine residues within glycoproteins that mediate inflammation [41].
The redox activity of metal complexes provides another strategic mechanism, particularly for complexes of iron, copper, and cobalt that can participate in Fenton-type reactions to generate reactive oxygen species [39]. This approach is especially valuable against bacterial pathogens that have developed resistance to conventional antibiotics through mechanisms such as drug efflux pump systems, plasmid-mediated gene transfer, and biofilm formation [39]. The multifactorial attack enabled by metal complexes makes it significantly more difficult for pathogens to develop resistance compared to single-target organic antibiotics.
Experimental Protocols and Methodologies
Protocol 1: Investigating Fe-S Cluster Assembly and Disruption
Objective: To assess the effect of metal compounds on iron-sulfur cluster assembly using Escherichia coli IscU, the scaffold protein on which Fe-S clusters are assembled [42].
Materials and Reagents:
- Uniformly nitrogen-15 labeled Escherichia coli IscU
- GaCl₃ or other metal salts for testing
- NMR buffer (20 mM HEPES, pH 7.4, 150 mM NaCl)
- Iron source (e.g., ammonium iron(II) sulfate hexahydrate)
- Sulfur source (e.g., L-cysteine)
- Cysteine desulfurase (IscS)
- Dithiothreitol (DTT) or other reducing agents
- Anaerobic chamber or sealed reaction vessels for oxygen-sensitive steps
Procedure:
- Protein Preparation: Express and purify uniformly ¹⁵N-labeled IscU from E. coli using established protocols [42].
- Sample Preparation: Prepare NMR samples containing 0.1-0.5 mM ¹⁵N-IscU in NMR buffer. Transfer to NMR tubes under anaerobic conditions.
- Cluster Assembly Reaction: Set up the Fe-S cluster assembly reaction containing:
- ¹⁵N-IscU (0.1 mM)
- IscS (0.01 mM)
- L-cysteine (1 mM)
- Ammonium iron(II) sulfate (0.1 mM)
- DTT (5 mM)
- Incubate at 37°C for 60 minutes under anaerobic conditions [42].
- Metal Treatment: Add GaCl₃ or test metal compounds at varying concentrations (0.01-1 mM) to separate samples.
- NMR Analysis: Acquire ¹H-¹⁵N HSQC spectra at 25°C using a high-field NMR spectrometer.
- Data Interpretation: Monitor chemical shift perturbations and peak intensity changes in IscU spectra. Cluster formation typically causes widespread signal broadening and disappearance, while cluster disruption may lead to signal reappearance.
Applications: This protocol allows evaluation of how metal-based compounds interfere with Fe-S cluster assembly, providing insights for developing antibacterial agents that target essential metalloenzyme pathways in pathogens.
Protocol 2: Assessing Antibacterial Activity of Transition Metal Complexes
Objective: To evaluate the efficacy of transition metal complexes against drug-resistant bacterial pathogens.
Materials and Reagents:
- Transition metal complexes (e.g., silver, copper, ruthenium, or iridium complexes)
- Bacterial strains (e.g., methicillin-resistant S. aureus, vancomycin-resistant Enterococcus)
- Mueller-Hinton broth or appropriate culture medium
- 96-well microtiter plates
- Positive control antibiotics (e.g., vancomycin, ampicillin)
- Phosphate-buffered saline (PBS)
- Resazurin solution or MTT for viability assessment
Procedure:
- Bacterial Culture: Grow bacterial strains to mid-log phase (OD₆₀₀ ≈ 0.5) in appropriate medium.
- Compound Preparation: Prepare serial dilutions of transition metal complexes in culture medium across a concentration range (typically 0.1-100 µg/mL).
- Inoculation: Add 100 µL of bacterial suspension (5 × 10⁵ CFU/mL) to each well of a 96-well plate containing 100 µL of diluted compounds.
- Incubation: Incubate plates at 37°C for 16-20 hours under appropriate atmospheric conditions.
- Viability Assessment: Add resazurin solution (20 µL of 0.15 mg/mL) to each well and incubate for 2-4 hours. Measure fluorescence (excitation 560 nm, emission 590 nm) [39].
- MIC Determination: Calculate minimum inhibitory concentration (MIC) as the lowest compound concentration that inhibits ≥90% of bacterial growth compared to untreated controls.
- Cytotoxicity Testing: Perform parallel assays with mammalian cell lines (e.g., HEK-293, HeLa) to determine selectivity indices.
Applications: This standardized protocol enables systematic evaluation of novel transition metal complexes against drug-resistant pathogens, facilitating structure-activity relationship studies and lead compound identification.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table: Key Research Reagent Solutions for Metalloprotein Studies
| Reagent/Material | Function/Application | Specific Examples | Technical Considerations |
|---|---|---|---|
| ¹⁵N-labeled Proteins | NMR studies of protein dynamics and metal interactions | Uniformly ¹⁵N-labeled IscU | Requires isotopic labeling during protein expression |
| Anaerobic Chambers | Oxygen-sensitive Fe-S cluster manipulations | Coy Laboratory Products | Maintain <1 ppm O₂ for cluster stability |
| EPR Spectroscopy | Characterization of paramagnetic Fe-S clusters | Bruker ELEXSYS systems | Low-temperature measurements (10-50 K) for cluster detection |
| Mössbauer Spectroscopy | Determination of Fe oxidation states and coordination | ⁵⁷Fe-enriched samples | Requires specialized instrumentation and isotope enrichment |
| Metal Complex Libraries | Screening for antimicrobial/antiparasitic activity | CO-ADD database compounds | Structure-diversity important for SAR studies |
| X-ray Crystallography | Structural determination of metal-protein complexes | Fe-S cluster enzymes | Rapid freezing for radiation-sensitive samples |
| UV-Vis Absorption | Monitoring cluster assembly/disassembly | [2Fe-2S] & [4Fe-4S] spectra | Characteristic features at ~420 nm for Fe-S clusters |
Integrated Signaling Pathways and Therapeutic Targeting
The intersection of Fe-S cluster biology and transition metal complex pharmacology creates unique opportunities for therapeutic intervention. Pathogens utilize Fe-S clusters in essential enzymes, making these systems attractive targets for antibacterial development. For instance, assimilatory sulfate reduction supplies prototrophic organisms with reduced sulfur required for biosynthesis of all sulfur-containing metabolites, and the enzymes involved (APS reductase and PAPS reductase) have no human homologs, rendering them attractive targets for novel antibacterial drugs [43]. Simultaneously, synthetic transition metal complexes can be designed to exploit the vulnerabilities of Fe-S cluster-containing proteins in pathogens while minimizing damage to human metalloenzymes.
Therapeutic Targeting Strategy: This diagram illustrates how transition metal complexes employ multiple mechanisms to target pathogen vulnerabilities, leading to diverse therapeutic applications.
The redox sensitivity of Fe-S clusters, while creating vulnerabilities in human disease states, also presents strategic opportunities for selective pathogen targeting. Recent studies have revealed that excess oxygen tensions disrupt multiple human pathways containing highly labile Fe-S cluster proteins, including purine metabolism, diphthamide synthesis, nucleotide excision repair, and electron transport chain activity [38]. This understanding informs the development of metal complexes that can generate localized oxidative stress to disrupt essential Fe-S cluster-dependent pathways in pathogens or cancerous cells.
Advanced characterization techniques are essential for studying these complex metallobiological systems. UV-Vis absorption, NMR, X-ray crystallography, EPR and Mössbauer spectroscopies, and electrochemical techniques provide complementary information about Fe-S cluster stoichiometry, oxidation states, and protein interactions [40]. These methods enable researchers to elucidate unique redox transitions of specific Fe-S clusters and understand how these transitions influence enzyme function and interactions, providing a foundation for rational drug design targeting metalloenzyme systems.
Self-Consistent Field (SCF) convergence represents a fundamental challenge in quantum chemical calculations, particularly for pathological systems such as open-shell transition metal complexes and conjugated radicals. The accuracy of methods like Density Functional Theory (DFT) is heavily dependent on achieving a fully converged SCF solution, as an unconverged wavefunction can lead to unreliable energies, geometries, and molecular properties. This application note details advanced guess strategy protocols—MORead, oxidized state convergence, and alternative guesses—framed within broader research on SCF block settings for pathological cases. These strategies are especially crucial for systems prevalent in drug discovery, including battery cathode materials, metal-containing enzymes, and redox-active pharmaceutical compounds, where an accurate description of electronic structure is essential for predicting properties and reactivity.
Theoretical Framework and Quantitative Data
The SCF Convergence Challenge
The SCF procedure iteratively searches for a self-consistent electron density. Convergence is typically assessed by the change in density between cycles, with the default criterion in packages like BAND being 1e-6 * sqrt(N_atoms) for Normal numerical quality [9]. For systems with strongly localized d or f electrons, standard DFT is affected by self-interaction errors (SIEs), leading to unphysical electron delocalization that impedes convergence and reduces accuracy. Extended Hubbard functionals (DFT+U+V) can mitigate these errors, providing a more reliable approach for redox-active materials and improving the description of oxidation states, which is critical for modeling processes in rechargeable batteries and electrochemical systems [44].
Quantitative Comparison of Advanced Guess Strategies
The following table summarizes the core characteristics, applications, and key parameters for the three advanced guess strategies discussed in this note.
Table 1: Comparison of Advanced SCF Initial Guess Strategies
| Strategy | Core Principle | Primary Application | Key Input Parameters | Expected Outcome |
|---|---|---|---|---|
| MORead | Utilizes pre-converged orbitals from a simpler, more robust calculation as the initial guess [1]. | Pathological systems where default guesses (e.g., PModel) fail; transition metal complexes [1]. | Path to orbital file (e.g., "bp-orbitals.gbw"); Level of theory of initial calculation (e.g., BP86/def2-SVP) [1]. |
Significantly improved starting point; Reduced SCF iterations; Higher convergence likelihood. |
| Oxidized State Convergence | Converges a closed-shell 1/2-electron oxidized state, using its orbitals as the guess for the target open-shell system [1]. | Troublesome open-shell systems, particularly radicals and transition metal complexes with challenging electronic configurations. | Charge and multiplicity of the oxidized/closed-shell state; Method for oxidizing state (e.g., ! Ion keyword in ORCA). |
Provides a stable, physically reasonable initial guess that can be smoothly mapped to the correct, more complex electronic structure. |
| Alternative Initial Guesses | Employs algorithms other than the default to generate the initial density or potential [1]. | Systems where the default guess (e.g., superposition of atomic densities - rho) leads to slow convergence or oscillations. |
Guess keyword (e.g., PAtom, Hueckel, HCore); InitialDensity (e.g., psi for atomic orbitals) [9] [1]. |
Alternative electron density distribution that may be closer to the true solution, breaking initial symmetry or degeneracy issues. |
Experimental Protocols
Protocol 1: MORead Strategy
This protocol is designed for systems where standard convergence methods fail.
1. Preliminary Calculation Setup
- Method Selection: Choose a computationally inexpensive and robust method, such as BP86 with a moderate basis set like def2-SVP [1].
- System Preparation: Use the identical molecular geometry intended for the high-level target calculation.
- SCF Settings: Employ default SCF convergence settings. The goal is to achieve a stable, converged solution, not high accuracy.
2. Execution and File Handling
- Run Calculation: Execute the preliminary single-point energy calculation.
- Orbital File Identification: Locate the output orbital file (e.g.,
.gbwin ORCA,.scfin BAND). - File Transfer: Ensure this orbital file is accessible for the subsequent high-level calculation.
3. High-Level Calculation with MORead
- Input Specification: In the input for the target (high-level) calculation, include the keyword for molecular orbital reading (e.g.,
! MOReadin ORCA) [1]. - Orbital Pathing: Specify the path to the preliminary orbital file using the appropriate block (e.g., in ORCA:
%moinp "bp-orbitals.gbw" end). - Execution: Run the target calculation. The SCF procedure will begin from the orbitals obtained in the preliminary calculation.
Protocol 2: Oxidized/Reduced State Convergence
This strategy is particularly effective for open-shell systems.
1. Oxidized/Reduced System Definition
- Electronic State Manipulation: Modify the input for your system to create a closed-shell or simpler open-shell configuration. This typically involves a 1- or 2-electron change in the system's charge [1].
- Geometry: Use the same molecular geometry as the target system.
2. Converging the Simpler State
- Method Selection: A standard DFT functional (e.g., PBE, B3LYP) is often sufficient.
- SCF Strategy: If convergence is difficult, apply the MORead protocol (Protocol 1) or use damping keywords (e.g.,
! SlowConv) to achieve SCF convergence for this manipulated state [1].
3. Orbital Mapping for Target System
- Input for Target System: Prepare the input for the original, problematic electronic state.
- Orbital Import: Use the
! MOReadkeyword and specify the path to the converged orbitals from the oxidized/reduced calculation [1]. - Execution: The calculation will start from the orbitals of the simpler state, which often provides a sufficiently good guess to converge the correct, more complex state.
Protocol 3: Application of Alternative Initial Guesses
This protocol addresses failures at the very beginning of the SCF process.
1. Initial Density Guess Selection
- Assessment: If the SCF fails immediately or oscillates wildly from the first iteration, the initial density guess is likely poor.
- Alternative Methods: Replace the default initial guess. Common alternatives include:
PAtom: Uses a superposition of atomic potentials.Hueckel: Uses a Hückel Hamiltonian for the initial guess.HCore: Uses the core Hamiltonian [1].
2. Input Modification
- Keyword Implementation: Add the chosen guess keyword to the input file (e.g., in ORCA:
! Guess PAtom). - BAND Specifics: In BAND, use the
InitialDensitykey within theConvergenceblock. Thepsioption constructs an initial eigensystem by occupying and orthonormalizing atomic orbitals, which can be a better starting point than the default sum of atomic densities (rho) [9].
3. Symmetry Breaking
- Initial Potential Splitting: To break initial alpha-beta degeneracy, use the
VSplitkeyword in theSCFblock (e.g.,VSplit 0.05adds a small constant to the beta spin potential at startup) [9]. - Maximum Spin: The
StartWithMaxSpinoption (default in BAND) is another strategy to break initial symmetry [9].
Workflow Visualization
The following diagram illustrates the logical decision process for selecting and applying these advanced guess strategies when facing SCF convergence failure.
Research Reagent Solutions
The following table details key computational "reagents" — the software, functionals, and algorithms essential for implementing the described protocols.
Table 2: Essential Computational Reagents for Advanced SCF Strategies
| Reagent / Tool | Type | Primary Function in Protocol | Example Usage |
|---|---|---|---|
| ORCA | Quantum Chemistry Software | Primary platform for running SCF calculations and implementing advanced guess strategies [1]. | ! MORead keyword to read initial orbitals; %moinp "guess.gbw" block to specify file. |
| BAND | DFT Software Package | Alternative platform with specific SCF control blocks and keywords [9]. | Convergence block with InitialDensity psi to use atomic orbitals for initial guess. |
| BP86/def2-SVP | DFT Functional / Basis Set | A robust, low-cost level of theory for generating initial guess orbitals via the MORead protocol [1]. | Initial calculation to produce a stable .gbw orbital file for a pathological system. |
| DFT+U+V | Advanced DFT Functional | Corrects self-interaction error for localized electrons, providing a better description of oxidation states and improving overall convergence landscape [44]. | ! DFT+U and ! DFT+V keywords to apply corrections to d/f orbitals and their hybridization. |
| PAtom, HCore, Hueckel | Alternative Guess Algorithms | Provides a different pathway to generate the initial electron density or potential when the default fails [1]. | ! Guess PAtom in the input file to invoke the superposition of atomic potentials. |
Overcoming Linear Dependencies in Large/Diffuse Basis Set Calculations
The use of large, diffuse basis sets is essential for achieving high accuracy in quantum chemical calculations, particularly for properties such as non-covalent interactions, excited states, and response properties. However, these basis sets introduce a significant computational challenge: linear dependence in the basis function representation. This problem arises when the overlap matrix between basis functions becomes ill-conditioned, meaning some eigenvalues approach zero, indicating that not all basis functions are linearly independent. Within the broader context of self-consistent field (SCF) methods for pathological convergence cases, linear dependencies can severely impede or even prevent SCF convergence. They destabilize the SCF procedure by making the orbital optimization problem ill-conditioned, leading to oscillatory or divergent behavior. This application note details protocols for diagnosing and overcoming these issues, enabling researchers to leverage the accuracy of diffuse basis sets without sacrificing computational robustness.
Quantitative Basis Set Analysis
The decision to use a diffuse basis set involves a critical trade-off between accuracy and numerical stability. The following table summarizes the performance characteristics of selected basis sets, illustrating this conundrum.
Table 1: Accuracy and Sparsity Trade-offs for Selected Basis Sets (ωB97X-V Functional)
| Basis Set | Total RMSD (kJ/mol) | NCI RMSD (kJ/mol) | Sparsity Impact |
|---|---|---|---|
| def2-SVP | 33.32 | 31.51 | High sparsity |
| def2-TZVP | 17.36 | 8.20 | Moderate sparsity |
| def2-TZVPPD | 16.40 | 2.45 | Very low sparsity |
| aug-cc-pVDZ | 26.75 | 4.83 | Very low sparsity |
| aug-cc-pVTZ | 17.01 | 2.50 | Very low sparsity |
Data adapted from Laqua et al. (2025) [45]. Total RMSD is the method+basis error for the entire ASCDB benchmark. NCI RMSD is the error for non-covalent interactions only. Sparsity impact refers to the effect on the one-particle density matrix.
The data shows that while augmented basis sets like def2-TZVPPD and aug-cc-pVTZ are essential for accurately modeling non-covalent interactions (achieving NCI RMSDs around 2.5 kJ/mol), they drastically reduce the sparsity of the one-particle density matrix. This "curse of sparsity" is a direct consequence of increased linear dependence and poses significant challenges for SCF convergence [45].
Core Computational Protocols
Protocol 1: Linear Dependency Diagnosis and Management
Principle: Identify and remove near-linear dependencies in the basis set to stabilize the SCF procedure.
Detailed Methodology:
- Compute the Overlap Matrix: Calculate the overlap matrix S for the atomic orbital basis set.
- Diagonalize the Overlap Matrix: Perform a canonical orthogonalization by solving the eigenvalue problem: S U = U λ, where λ is the diagonal matrix of eigenvalues.
- Set a Tolerance Threshold: Define a tolerance for the smallest allowable eigenvalue. A common starting point is 1e-7 [46]. Eigenvalues below this threshold indicate linear dependencies.
- Remove Dependent Functions: Construct a transformed basis by excluding the eigenvectors (columns of U) corresponding to eigenvalues below the tolerance threshold.
- Proceed with SCF: Use the transformed, linearly independent basis for the SCF calculation.
Most quantum chemistry packages automate this process. Key is adjusting the tolerance parameter:
- Tighter tolerance (e.g., 1e-6): Removes more functions, increases stability but risks losing chemical accuracy.
- Looser tolerance (e.g., 1e-8): Retains more functions, improving potential accuracy but may not resolve severe convergence issues.
Protocol 2: SCF Stabilization for Ill-Conditioned Systems
Principle: Use specialized SCF algorithms and parameters to guide convergence when the basis set is near-linear dependent.
Detailed Methodology: Implement the following settings in your SCF input block, as supported by codes like ADF, ORCA, and Psi4 [28] [18] [46].
Table 2: Key SCF Settings for Pathological Convergence
| Setting | Recommended Value | Function |
|---|---|---|
| Overlap Threshold | S_TOLERANCE 1e-7 [46] |
Removes linear dependencies based on overlap eigenvalue. |
| DIIS Vectors | DIIS N 12 [28] |
Increases history for better extrapolation; crucial for difficult cases. |
| Level Shifting | Lshift 0.5 [28] |
Shifts virtual orbitals, damping oscillations (enables OldSCF in ADF). |
| Damping | DAMPING_PERCENTAGE 20 [46] |
Mixes a percentage of old density to dampen oscillations. |
| SCF Accelerator | AccelerationMethod LISTb [28] or SCF_INITIAL_ACCELERATOR ADIIS [46] |
Uses robust algorithms less prone to divergence. |
Workflow Integration: The relationship between diagnosing linear dependencies and applying SCF stabilization techniques is a sequential, decision-based process.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Managing Linear Dependencies
| Tool / Reagent | Function / Purpose | Example Implementation |
|---|---|---|
| Overlap Eigenvalue Analysis | Diagnoses the severity of linear dependencies by identifying near-zero eigenvalues. | S_TOLERANCE in Psi4 [46]; Canonical orthogonalization in codes like ORCA. |
| Dunning's aug-cc-pVXZ | Provides a systematic series of correlation-consistent basis sets with diffuse functions for pursuing the complete basis set (CBS) limit. | basis aug-cc-pVTZ [47] [45] |
| Karlsruhe def2-XVPPD | Offers a family of polarized, diffuse basis sets balancing accuracy and computational cost. | basis def2-TZVPPD [45] [48] |
| DIIS / LIST Accelerators | Extrapolates Fock matrices from previous iterations to accelerate SCF convergence. | DIIS N 12 [28]; AccelerationMethod LISTb [28]. |
| Damping & Level Shift | Numerical stabilizers that dampen charge oscillations, a common symptom of linear dependencies. | DAMPING_PERCENTAGE 20 [46]; Lshift 0.5 [28]. |
| Complementary Auxiliary Basis Sets (CABS) | A potential solution that can improve accuracy with smaller primary basis sets, mitigating the linear dependence problem [45]. | Used in explicitly correlated (F12) methods. |
Advanced Application: Excited States and Density Fitting
Linear dependencies pose a particular challenge for excited state and linear response calculations, where large, diffuse basis sets are mandatory for accurate polarizabilities and excitation energies [47]. In such cases, combining the above SCF stabilization protocols with density fitting (DF) techniques is highly effective.
Protocol for DF-SCF:
- Employ a Robust Guess: Use
DF_SCF_GUESS TRUEto converge orbitals with a fitted density before switching to exact integrals [46]. - Use a Appropriate Auxiliary Basis: Specify a DF basis tailored for your diffuse primary basis (
DF_BASIS_SCF). - Consider a Basis Set Guess: For extremely problematic cases, project the solution from a smaller, more stable basis set (
BASIS_GUESS TRUE) [46].
For ΔSCF excited state calculations, which can access states like double excitations that are challenging for TDDFT, ensuring a stable, oscillation-free ground state SCF is a critical prerequisite [49]. The protocols outlined herein provide a foundation for achieving this stability.
Validating Solutions and Comparing Method Efficacy Across Systems
In computational chemistry, particularly in drug development where accurate prediction of molecular properties and reaction pathways is paramount, obtaining a self-consistent field (SCF) solution is only the first step. Two critical, and often overlooked, diagnostic procedures are essential for ensuring the physical meaningfulness of the result: SCF stability analysis and spin contamination checks. A converged SCF wavefunction may correspond to a saddle point rather than a true minimum, or be contaminated by higher spin states, leading to significant errors in computed energies, geometries, and spin densities. These errors can profoundly impact predictions of drug-receptor binding, reaction mechanisms, and spectroscopic properties. This application note provides detailed protocols for identifying and correcting these issues, framed within broader research on SCF convergence for pathologically challenging systems, such as open-shell transition metal complexes and diradicals common in catalytic drug synthesis.
SCF Stability Analysis: Theory and Protocol
Theoretical Foundation
The SCF procedure locates a stationary point in the energy landscape with respect to changes in molecular orbital coefficients. A stability analysis evaluates the electronic Hessian (second derivative matrix) at this point. A positive-definite Hessian (all eigenvalues positive) indicates a local energy minimum, while negative eigenvalues reveal an unstable saddle point, from which the energy can be lowered by breaking the wavefunction's symmetry or constraints [50] [51] [52]. Common instabilities include:
- Restricted → Unstable (RHF → UHF): A restricted solution (RHF) is unstable towards breaking spin symmetry to form an unrestricted solution (UHF), often occurring in stretched bonds or diradicals [50] [52].
- Real → Complex Instability: A solution using real orbitals is unstable towards a lower-energy solution requiring complex orbitals [51].
- Unrestricted → Unrestricted Instability: A UHF solution is unstable towards a different, lower-energy UHF solution [52].
Detailed Experimental Protocol
This protocol outlines the steps for performing an internal stability analysis and acting on its results, using typical keywords for ORCA [50] [52] and Q-Chem [51].
Table 1: Key Settings for SCF Stability Analysis in Different Software Packages
| Setting | ORCA Input Example | Q-Chem Input Example | Purpose and Notes |
|---|---|---|---|
| Activate Analysis | STABPerform true in %scf block |
INTERNAL_STABILITY = TRUE |
Instructs the program to perform stability analysis after SCF convergence. |
| Number of Roots | STABNRoots 3 |
INTERNAL_STABILITY_ROOTS = 2 |
Number of lowest Hessian eigenvalues to find. 2-3 is usually sufficient [50] [51]. |
| Auto-restart | STABRestartUHFifUnstable true |
INTERNAL_STABILITY_ITER = 1 |
Automatically restarts SCF from a perturbed guess if unstable. Q-Chem can perform multiple macro-iterations [51]. |
| Hessian Evaluation | N/A (Analytical default) | FD_MAT_VEC_PROD = FALSE |
Use finite-difference for Hessian if analytical is unavailable (e.g., for certain functionals) [51]. |
| Convergence | STABRTol 0.0001 |
INTERNAL_STABILITY_CONV = 4 |
Convergence tolerance for the Davidson solver (10⁻ⁿ for residual norm) [50] [51]. |
Procedure:
- Initial SCF Calculation: Converge the initial SCF calculation for your system using a standard method (e.g., DFT/B3LYP). The stability analysis is typically run as a post-SCF procedure.
- Configure and Execute Stability Analysis: Add the appropriate keywords from Table 1 to your input file. For ORCA, this is typically done within the
%scfblock [50] [52]. For Q-Chem, the relevant$remvariables are used [51]. - Interpret Results: Examine the output for the eigenvalues of the electronic Hessian.
- Stable Solution: All eigenvalues are positive. The wavefunction is at a local minimum.
- Unstable Solution: One or more negative eigenvalues are found. The solution is a saddle point and should not be used.
- Follow-up Actions for Unstable Solutions:
- Automated Correction: If
STABRestartUHFifUnstable(ORCA) orINTERNAL_STABILITY_ITER(Q-Chem) is active, the program will automatically generate a new guess by displacing the orbitals along the direction of the unstable mode and restart the SCF [51] [52]. - Manual Investigation: If automatic correction fails or is not enabled, the result mandates a change in the initial SCF strategy. This often means switching from a restricted (RKS/RHF) to an unrestricted (UKS/UHF) formalism, or using a different initial guess (e.g.,
Guess PModelinstead ofGuess HCorein ORCA) [1] [52].
- Automated Correction: If
The following workflow diagram summarizes the logical process of performing stability analysis and the decision points based on its outcome.
Spin Contamination: Theory and Protocol
Theoretical Foundation
Spin contamination is a specific pathology of unrestricted wavefunctions (UHF, UDFT). The wavefunction is no longer an eigenfunction of the total spin operator ( \hat{S}^2 ) and becomes artificially contaminated by wavefunctions of higher spin states [53] [54]. This occurs because alpha and beta electrons are allowed to occupy different spatial orbitals without constraint, introducing an unphysical mixture of spin states into the single-determinant wavefunction.
While spin contamination can sometimes artificially lower the energy by providing more variational freedom, it more often raises the energy and adversely affects computed properties. It is a significant source of error in:
- Energy differences (e.g., reaction barriers, singlet-triplet gaps)
- Molecular geometries and bond lengths
- Population analyses and, most critically, spin densities [53] [54]
Spin contamination is generally less severe in pure DFT calculations than in hybrid DFT or pure Hartree-Fock due to the different nature of the exchange-correlation functional, but it must always be checked when using unrestricted methods [53] [54].
Detailed Experimental Protocol
This protocol describes how to quantify spin contamination and implement strategies to mitigate it.
Table 2: Assessment and Mitigation of Spin Contamination
| Aspect | Procedure and Interpretation | Remedial Actions |
|---|---|---|
| Quantification | Compute the expectation value ( \langle S^2 \rangle ) from the output. For a doublet (one unpaired electron), the exact value is ( s(s+1) = 0.75 ). Compare the computed value to the exact value. | N/A |
| Threshold | A deviation of less than 10% is often considered acceptable for organic molecules (e.g., < 0.825 for a doublet). Larger deviations indicate significant contamination [53] [54]. | N/A |
| Mitigation Strategy 1 | Restricted Open-Shell (ROHF/ROKS): Eliminates spin contamination by enforcing a single set of orbitals for doubly-occupied and open-shell spaces. Disadvantages: Loss of spin polarization, more computationally expensive, and orbital energies lack rigorous meaning [53] [54]. | Use keywords for restricted open-shell calculations (e.g., ROHF in Gaussian). |
| Mitigation Strategy 2 | Alternative DFT Functional: Reduce the amount of exact Hartree-Fock exchange in hybrid functionals, as HF exchange is a primary driver of spin contamination in DFT [54]. | Switch from a hybrid (e.g., B3LYP) to a pure (e.g., BP86) or low-HF-exchange functional. |
| Mitigation Strategy 3 | Spin-Projection Methods (e.g., PUHF): Projects out the contaminated parts of the wavefunction after convergence. Note: The orbitals are not re-optimized for the projected state, which can lead to inconsistencies [53]. | Use specialized keywords (e.g., PMP2 in Gaussian), but be aware of the limitations. |
Procedure:
- Perform an Unrestricted Calculation: Run a standard UHF or UKS calculation.
- Check the ( \langle S^2 \rangle ) Value: Locate the computed ( \langle S^2 \rangle ) value in the output file. Most quantum chemistry software (ORCA, Gaussian, Q-Chem, etc.) prints this value during the SCF procedure.
- Calculate and Assess Deviation:
- Calculate the exact value: ( s = n/2 ), where ( n ) is the number of unpaired electrons. The exact ( \langle S^2 \rangle ) is ( s(s+1) ).
- For a triplet (n=2): ( s=1 ), exact value = 2.00.
- For a doublet (n=1): ( s=0.5 ), exact value = 0.75.
- Compute the percentage deviation: ( \text{Deviation} = \frac{ \langle S^2 \rangle{\text{computed}} - \langle S^2 \rangle{\text{exact}} }{ \langle S^2 \rangle_{\text{exact}} } \times 100\% ).
- Implement Mitigation Strategies: If the contamination exceeds acceptable limits (e.g., >10%), proceed with the strategies outlined in Table 2. The most robust solution is often to switch to a Restricted Open-Shell (ROHF/ROKS) formalism, despite its computational cost and other limitations [53].
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Key Computational Tools and Methods for Diagnosing SCF Pathologies
| Tool/Solution | Function | Example Use-Case |
|---|---|---|
| SCF Stability Analysis | Diagnoses if a converged wavefunction is at a true minimum or an unstable saddle point. | Identifying a falsely stable restricted (RHF) solution for a diradical molecule, leading to an incorrect unrestricted (UHF) solution with lower energy. |
| ( \langle S^2 \rangle ) Diagnostic | Quantifies the amount of spin contamination in an unrestricted wavefunction. | Detecting unphysical spin densities in a transition metal catalyst radical intermediate. |
| Restricted Open-Shell (ROHF/ROKS) | Provides a spin-pure wavefunction for open-shell systems, eliminating spin contamination. | Calculating reliable spin densities and energies for organic radical reaction pathways. |
| Second-Order SCF (SOSCF) | Robust SCF converger that can help reach convergence in difficult cases, often used with KDIIS [1]. |
Converging the SCF for open-shell transition metal complexes where the default DIIS algorithm fails. |
| Trust Radius Augmented Hessian (TRAH) | A robust, albeit more expensive, second-order SCF convergence method in ORCA [1]. | Automatically activated in ORCA when the default DIIS procedure struggles; can be forced with !TRAH. |
| Damping / Level Shifting | Stabilizes the initial SCF iterations by mixing old and new densities (Damping) or shifting virtual orbital energies (Shift) [1]. |
Quenching oscillations in the SCF energy during the first iterations of a calculation on a metal cluster. |
| MORead Guess | Uses orbitals from a previous, simpler calculation (e.g., BP86) as a guess for a more challenging one (e.g., a hybrid functional) [1]. | "Bootstrapping" convergence for a pathological system by first converging a calculation with a pure functional and small basis set. |
Integrated Workflow for Pathological Convergence Cases
For researchers dealing with pathologically challenging systems, such as open-shell transition metal compounds, metal clusters, or molecules with stretched bonds, an integrated diagnostic and corrective workflow is essential. The following protocol combines stability and spin diagnostics with advanced SCF convergence techniques.
Procedure:
- Initial Setup and Simple Guess: Begin with a coarse integration grid and a modest basis set if possible. Use a robust initial guess like
PAtomorHueckelin ORCA, which can be more effective than the defaultPModelfor difficult systems [1]. - Converge a Preliminary SCF: Attempt convergence using a pure GGA functional (e.g., BP86) which is less prone to convergence issues and spin contamination than hybrid functionals. Employ convergence aids like
!SlowConvor damping if necessary [1]. - Refine Calculation: Use the converged orbitals from step 2 (
!MORead) as the guess for the target level of theory (e.g., a hybrid functional and a larger basis set). - Apply Advanced SCF Settings: If convergence stalls, implement more robust algorithms. In ORCA, a combination of
!KDIISand!SOSCFis often effective. For truly pathological cases, increase the DIIS subspace (DIISMaxEq 15-40) and the maximum iterations (MaxIter 500-1500) [1]. - Execute Diagnostic Suite:
- Perform SCF Stability Analysis: Follow the protocol in Section 2.2. If unstable, allow the program to restart or manually switch to an unrestricted formalism.
- Check for Spin Contamination: If an unrestricted solution was used, follow the protocol in Section 3.2 to check ( \langle S^2 \rangle ).
- Finalize Stable, Spin-Pure Solution: If spin contamination is high, consider switching to a Restricted Open-Shell (ROKS) calculation or a functional with less exact exchange to obtain a physically meaningful result [53] [54].
Self-Consistent Field (SCF) convergence is a fundamental challenge in computational chemistry, with the selection of convergence criteria directly impacting the reliability of results and consumption of computational resources. This challenge is particularly acute for pathological systems such as open-shell transition metal complexes, metal clusters, and molecules with multireference character or at pathological geometries. Achieving convergence in these cases often requires specialized SCF block settings and a deep understanding of the trade-offs involved. This Application Note provides detailed protocols for selecting SCF convergence criteria and algorithms, specifically framed within research on pathological convergence cases, to enable researchers to make informed decisions that balance accuracy and computational cost.
Understanding SCF Convergence Tolerances
Core Convergence Criteria
SCF convergence is typically assessed through multiple, interdependent criteria. ORCA, for example, monitors several key parameters, and a calculation is considered converged only when all specified tolerances are met, depending on the ConvCheckMode setting [18]. The most commonly monitored criteria and their interpretations are:
TolE: The change in the total energy between two consecutive SCF cycles. Convergence requires this change to fall below the threshold.TolMaxP: The maximum change in any element of the density matrix.TolRMSP: The root-mean-square change in the density matrix.TolErr: The error vector within the DIIS (Direct Inversion in the Iterative Subspace) acceleration algorithm.TolG: The maximum element of the orbital gradient, which should be zero at a converged solution.
Standard Tolerance Presets
Quantum chemistry packages provide preset combinations of these tolerances to simplify input while ensuring self-consistency. The table below summarizes the standard tolerance presets available in ORCA, which exemplify the spectrum of accuracy from cursory to extreme [18].
Table 1: Standard SCF Convergence Tolerance Presets in ORCA (Selected Examples) [18]
| Preset | TolE (Hartree) | TolMaxP | TolRMSP | TolErr | Typical Use Case |
|---|---|---|---|---|---|
| SloppySCF | 3e-5 | 1e-4 | 1e-5 | 1e-4 | Initial geometry scans, qualitative MO analysis |
| LooseSCF | 1e-5 | 1e-3 | 1e-4 | 5e-4 | Preliminary geometry optimizations |
| MediumSCF | 1e-6 | 1e-5 | 1e-6 | 1e-5 | Default for single-point energies |
| StrongSCF | 3e-7 | 3e-6 | 1e-7 | 3e-6 | Default for geometry optimizations, transition metal systems |
| TightSCF | 1e-8 | 1e-7 | 5e-9 | 5e-7 | Final single-point energies, property calculations |
| VeryTightSCF | 1e-9 | 1e-8 | 1e-9 | 1e-8 | Challenging property calculations (e.g., NMR) |
| ExtremeSCF | 1e-14 | 1e-14 | 1e-14 | 1e-14 | Benchmarking, near-machine-precision studies |
For context, other software like ADF/BAND define their convergence criterion based on the integral of the squared difference between input and output densities, which is then normalized by system size (e.g., default of 1e-6 * sqrt(N_atoms) for Normal numerical quality) [9].
Experimental Protocols for Pathological Systems
Protocol 1: Systematic SCF Convergence for Stubborn Molecules
This protocol is designed for systems where standard DIIS procedures fail, often indicated by large oscillations in the early SCF iterations or convergence stalling at a high error.
Workflow Overview
Step-by-Step Methodology
Initial Stabilization with Damping
- Action: Apply damping to control large oscillations in initial SCF cycles.
- ORCA Input:
- Rationale: The
SlowConvkeyword increases damping, aiding convergence for systems with strong coupling or near-degeneracies [1]. IncreasingMaxIterprovides more time for convergence.
Advanced Algorithm Selection
- Action: If damping alone fails, employ more robust SCF convergers.
- ORCA Input: Alternative for DIIS failure:
- Rationale:
TRAH(Trust Radius Augmented Hessian) is a robust second-order method automatically activated in ORCA if DIIS struggles [1].KDIISwithSOSCFcan be faster for some systems, butSOSCFmay require a delayed start for open-shell cases [1].
Orbital Guess Refinement
- Action: Use a converged wavefunction from a simpler method or stable initial guess.
- ORCA Input: Alternative for bad guesses:
- Rationale: Reading a pre-converged set of molecular orbitals (
MORead) provides a high-quality starting point [1]. Changing theGuesscan help if the defaultPModelguess is unsuitable.
Persistent Convergence for Pathological Cases
- Action: For extremely difficult systems (e.g., iron-sulfur clusters), use specialized settings.
- ORCA Input:
- Rationale: A large
DIISMaxEq(15-40) provides more history for extrapolation. Settingdirectresetfreq 1rebuilds the Fock matrix every cycle, eliminating numerical noise that hinders convergence, despite being computationally expensive [1].
Protocol 2: Handling Specific Pathologies
Different types of difficult systems require tailored approaches.
- Open-Shell Transition Metal Complexes: Use
! SlowConvor! VerySlowConvwith potential level shifting (%scf Shift 0.1; end). For UHF/UKS, consider enablingSOSCFwith a cautious startup threshold [1]. - Conjugated Radical Anions with Diffuse Functions: These can suffer from linear dependence and poor convergence. Using a full Fock rebuild and an early-starting
SOSCFcan help [1]. - Systems at Pathological Geometries: When atoms are far apart, the initial guess can be problematic. Check the geometry reasonability. Try converging a different, simpler electronic state (e.g., cation or high-spin state) and use its orbitals as a guess for the target state [1] [5]. Using a more robust guess like
Guess GWHhas been reported to help in such cases [5].
The Scientist's Toolkit: Research Reagent Solutions
This section details key software tools, algorithms, and input parameters essential for tackling SCF convergence problems.
Table 2: Essential "Reagents" for SCF Convergence Research
| Item / Keyword | Function / Purpose | Application Context |
|---|---|---|
| DIIS (Default) | Fast convergence accelerator; extrapolates Fock matrices from previous cycles. | Standard for most well-behaved, closed-shell molecules. |
| TRAH | Robust second-order convergence algorithm; more reliable but slower than DIIS. | Automatic fallback in ORCA when DIIS struggles; recommended for guaranteed convergence [1]. |
| KDIIS | An alternative DIIS algorithm that can be faster for some systems. | Can be combined with SOSCF for efficient convergence in difficult cases [1]. |
| SOSCF | Second-Order SCF; uses exact Hessian information for rapid final convergence. | Activated once orbital gradient is small. Not always suitable for open-shell systems [1]. |
| SlowConv / VerySlowConv | Applies stronger damping to control oscillatory behavior in early SCF cycles. | Essential for open-shell transition metal complexes and other systems with large initial fluctuations [1]. |
| MORead | Reads initial molecular orbitals from a previous calculation. | Provides a high-quality guess, crucial for continuing calculations or using a stable guess from a simpler method [1]. |
| Guess | Generates the initial guess for the SCF procedure (e.g., PAtom, HCore). |
Alternative guesses can be more stable than the default for systems with unusual electronic structures [1]. |
| SCF Convergence Presets | Pre-defined sets of tolerances for energy, density, and gradient. | Simplifies input while ensuring consistency (e.g., TightSCF for high accuracy) [18]. |
Case Study: Troubleshooting a Non-Converging System
Problem: An SCF calculation for a large, open-shell transition metal complex fails to converge after 125 default cycles, showing oscillatory behavior.
Diagnosis and Solution Path: The oscillatory behavior suggests a need for damping and a more robust algorithm. The following troubleshooting decision tree outlines the recommended escalation path.
Application of Protocol:
- Initial Stabilization: The calculation is restarted with
! SlowConvandMaxIter 500[1]. - Algorithm Switch: If oscillations persist,
! TRAHis explicitly requested to leverage its second-order convergence guarantees [1]. - Guess Refinement: If convergence is still slow, a previously converged wavefunction from a lower level of theory (e.g., BP86/def2-SVP) is read in using
! MOReadto provide a better starting point [1]. - Final Effort: For ultimate pathological cases, the expert settings with a large DIIS subspace and frequent Fock rebuilds are deployed [1].
This systematic approach, moving from standard to specialized tools, maximizes the likelihood of achieving convergence while maintaining an awareness of the computational cost of each step.
Self-Consistent Field (SCF) convergence presents a significant challenge in quantum chemical calculations, particularly for pathological systems such as open-shell transition metal complexes and organic radical anions. These systems are characterized by complex electronic structures with near-degenerate orbitals, strong correlation effects, and inherent multi-reference character, which often lead to oscillatory behavior or complete failure of standard SCF procedures. The development and benchmarking of robust SCF algorithms are therefore crucial for advancing computational research in catalysis, materials science, and drug development where such molecular architectures are prevalent. This application note provides a structured framework for evaluating SCF algorithm performance, with specific protocols for handling these notoriously difficult cases, enabling researchers to make informed methodological choices.
Quantitative Benchmarking Data
Performance of Density Functionals for Disulfide Radical Anions
The performance of various density functional classes for describing disulfide radical anions (exemplified by dimethyldisulfide) is summarized below, with adiabatic electron affinity (AEA) and inter-sulfur distance (dSS) as key metrics [55].
Table 1: Performance of Density Functional Types for 2-Center-3-Electron Systems (e.g., Dimethyldisulfide Radical Anion)
| Functional Class | Representative Functionals | AEA Accuracy | dSS Accuracy | Recommended for 2c-3e Systems? |
|---|---|---|---|---|
| Global Hybrid GGA | B3LYP, B3P86 | Poor | Poor (Serious overestimation) | No |
| Global Hybrid Meta-GGA | B1B95, BMK | Good | Good | Yes |
| Range-Separated Hybrid (RSH) | ωB97X, CAM-B3LYP | Good | Good | Yes |
| Double Hybrid | B2PLYP(D) | Good | Good | Yes |
| Half-and-Half Hybrid | BH&HLYP | Moderate | Moderate (Overestimation ~0.09 Å) | Yes, but outdated |
Performance of Methods for Transition Metal Spin-State Energetics
Benchmarking against the experimentally derived SSE17 dataset reveals the performance of various quantum chemistry methods for predicting spin-state energetics in transition metal complexes [56].
Table 2: Benchmark Performance for Transition Metal Complex Spin-State Energetics (SSE17 Set)
| Method | Method Class | Mean Absolute Error (kcal mol⁻¹) | Maximum Error (kcal mol⁻¹) | Recommended Use |
|---|---|---|---|---|
| CCSD(T) | Wave Function Theory | 1.5 | -3.5 | High-accuracy reference |
| PWPB95-D3(BJ) | Double Hybrid DFT | < 3.0 | < 6.0 | Best-performing DFT |
| B2PLYP-D3(BJ) | Double Hybrid DFT | < 3.0 | < 6.0 | Best-performing DFT |
| B3LYP*-D3(BJ) | Global Hybrid DFT | 5-7 | > 10 | Not recommended |
| TPSSh-D3(BJ) | Meta-GGA Hybrid DFT | 5-7 | > 10 | Not recommended |
| CASPT2 | Multireference | > 1.5 | > -3.5 | Good alternative to CCSD(T) |
Experimental Protocols
Protocol 1: Benchmarking SCF Algorithms for Pathological Geometries
Objective: To systematically evaluate the efficacy of different SCF algorithms and settings in achieving convergence for molecules with pathological, high-energy geometries often encountered during potential energy surface scans [5].
Methodology:
System Preparation: Select or generate molecular geometries that are known to be problematic, such as distorted structures with atoms at extreme separations or incipient bond dissociation. An example input structure for a radical anion system (charge = -1, multiplicity = 1) is provided below [5]:
Initial Calculation Setup:
- Software: PSI4, ORCA, or Gaussian.
- Method: HF or a pure GGA functional (e.g., BP86).
- Basis Set: Start with a moderate basis set (e.g., 6-31G* or def2-SVP).
- SCF Settings: Use default settings and
MaxIter 200.
Systematic Algorithm Testing: Execute calculations using a series of SCF strategies, recording the number of iterations to convergence and success/failure. Key strategies to test include [1]:
- Default DIIS
- Damping: Set
DAMPING_PERCENTAGE 20in PSI4 or useSlowConvin ORCA. - Second-Order Methods: Use
TRAHin ORCA orNRSCF. - Improved Guess: Employ the
GWH(Gauss-Weinhold) guess in PSI4 orPAtom/HCorein ORCA instead of the default. - Integral Accuracy: Tighten integral thresholds (
ints_tolerance 1.0E-16in PSI4).
Data Analysis: Compare the performance of each algorithm based on the convergence rate, number of iterations, and final energy stability. Strategies that fail on the target system should be tested on a simplified system (e.g., smaller basis set, different spin state) to check for transferability [5].
Protocol 2: SCF Convergence for Open-Shell Transition Metal Complexes
Objective: To obtain a converged SCF solution for challenging open-shell transition metal complexes, which often exhibit severe convergence issues due to dense orbital manifolds and near-degeneracies [1].
Methodology:
Initial Attempt with Robust Defaults:
- Software: ORCA is highly recommended for such systems.
- Method: Use a functional like PBE0 or TPSSh.
- Basis Set: def2-SVP for metals, def2-SVP for light atoms.
- SCF Settings: Rely on the modern default in ORCA 5.0+, which automatically activates the Trust Radius Augmented Hessian (TRAH) algorithm if the standard DIIS struggles [1]. Use
TightSCFconvergence criteria.
Troubleshooting Non-Convergence: If the default procedure fails, employ a graded strategy:
- Step 1: Increase the maximum number of iterations (
%scf MaxIter 500 end). - Step 2: Use dedicated keywords for difficult cases:
SlowConvorVerySlowConvto increase damping [1]. - Step 3: Manually configure the TRAH algorithm if it struggles with its default settings [1]:
- Step 4: For truly pathological cases (e.g., iron-sulfur clusters), use advanced DIIS controls [1]:
- Step 1: Increase the maximum number of iterations (
Alternative Pathways: If the above fails, try converging the SCF for a different, easier-to-converge electronic state (e.g., a closed-shell oxidized state) and use its orbitals as a guess for the target state via
MORead[1].
Protocol 3: Accurate Calculation of Redox Properties for Radical Anions
Objective: To compute accurate ground- and excited-state redox potentials for organic photoredox catalysts and radical anions, which requires a balanced methodological approach [57].
Methodology:
Geometry Optimizations:
- Software: Gaussian 16.
- Method: Select a functional benchmarked for the property of interest. For excited-state redox potentials, PBE0-D3BJ/6-311+G(d,p) is recommended for S1 states, and ωB97X/6-311+G(d,p) or BHandH/6-311+G(d,p) for T1 states [57].
- Basis Set: 6-311+G(d,p) or aug-cc-pVDZ.
- Solvation: Use an implicit solvation model (e.g., IEF-PCM with parameters for acetonitrile, MeCN).
- SCF: Employ tight convergence criteria (e.g.,
SCF=Tightin Gaussian).
Single-Point Energy and Property Calculation:
- Functional/Basis: Consistent with optimization or a larger basis (e.g., cc-pVTZ).
- Calculation Types:
- Ground State (S0): Standard DFT calculation.
- Triplet State (T1): DFT with
stable=optto check stability. - Singlet Excited State (S1): Time-Dependent DFT (TD-DFT).
- Properties: Compute the free energy in solution for the neutral, radical anion, and radical cation species.
Redox Potential Calculation: Calculate the adiabatic electron affinity (AEA) and oxidation potential using the free energy differences between the optimized redox partners, referenced to a standard electrode (e.g., SCE) [55] [57].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for SCF Convergence and Benchmarking
| Tool / Reagent | Function / Purpose | Example Use Case |
|---|---|---|
| Double-Hybrid Functionals (PWPB95, B2PLYP) | Superior description of spin-state energetics and correlation [56]. | Benchmarking spin-state energy splittings in Fe(II) complexes [56]. |
| Range-Separated Hybrids (ωB97X, CAM-B3LYP) | Mitigate self-interaction error for charge-transfer and radical systems [55] [57]. | Calculating accurate electron affinities and excitation energies for radical anions [55]. |
| TRAH Algorithm | Robust second-order SCF converger for pathological cases [1]. | Converging open-shell transition metal complexes where DIIS fails [1]. |
| Damping & SlowConv | Stabilizes SCF iterations by mixing old and new densities [1]. | Handling oscillatory convergence in the initial SCF cycles [1]. |
| DIISMaxEq & directresetfreq | Advanced DIIS controls for numerical stability in difficult cases [1]. | Converging large, metallic clusters like iron-sulfur proteins [1]. |
| aug-cc-pVTZ Basis Set | Augmented, correlation-consistent basis for accurate anion and property calculations [55]. | Reference calculations for disulfide radical anion geometries and AEAs [55]. |
Self-Consistent Field (SCF) convergence represents a fundamental challenge in quantum chemistry simulations, particularly for pathological cases including open-shell transition metal complexes, radical anions with diffuse functions, and large metal clusters. Achieving convergence in these systems is critical for obtaining reliable energies, properties, and geometries in computational chemistry workflows, especially in drug development where accurate prediction of molecular behavior is paramount. This application note provides a structured comparison of three prominent quantum chemistry packages—ORCA, Q-Chem, and ADF—focusing on their specialized capabilities, protocols, and parameterizations for addressing challenging SCF convergence scenarios. The analysis presented herein is framed within broader thesis research on advanced SCF block configurations for pathological cases, providing practicing researchers with practical methodologies and decision frameworks for selecting and implementing appropriate software solutions.
Core Algorithmic Approaches
Each software package implements a distinct architectural philosophy for SCF convergence, employing different algorithms and fallback strategies when initial methods fail.
ORCA utilizes a sophisticated multi-algorithm approach that begins with efficient DIIS (Direct Inversion in the Iterative Subspace) procedures and automatically activates more robust methods upon detection of convergence difficulties. Its Trust Radius Augmented Hessian (TRAH) method provides a robust second-order convergence algorithm that automatically engages when the standard DIIS-based converger struggles [1]. For particularly challenging cases, ORCA implements specialized keywords (SlowConv, VerySlowConv) that modify damping parameters to control large fluctuations in early SCF iterations [1].
Q-Chem employs a highly modular SCF architecture offering multiple algorithm options selectable via the SCF_ALGORITHM variable. While DIIS remains the default for most calculations, Q-Chem's distinctive strength lies in its Geometric Direct Minimization (GDM) algorithm, which properly accounts for the hyperspherical geometry of orbital rotation space, providing exceptional robustness for difficult cases [10]. The software also implements adaptive switching between algorithms, allowing calculations to begin with DIIS for efficiency before transitioning to GDM when nearing convergence.
ADF (Amsterdam Density Functional), while less extensively documented in the available search results for specific SCF convergence techniques, employs a density-functional theory approach specifically designed for molecular calculations. As part of the Software for Chemistry & Materials (SCM) package, ADF specializes in handling challenging systems including transition metals and heavy elements [58].
Table 1: Core SCF Algorithm Profiles
| Software | Primary Algorithm | Secondary/Fallback Methods | Specialized Convergence Keywords |
|---|---|---|---|
| ORCA | DIIS with SOSCF | TRAH, KDIIS, NRSCF, AHSCF | SlowConv, VerySlowConv, TightSCF |
| Q-Chem | DIIS | GDM, ADIIS, DM, RCA | SCF_ALGORITHM options, DIIS_SUBSPACE_SIZE |
| ADF | DFT-specific procedures | Not specified in results | Not detailed in available sources |
Convergence Tolerance Hierarchies
Each package implements customizable convergence tolerance hierarchies, allowing researchers to balance computational efficiency with accuracy based on their specific needs.
ORCA provides predefined convergence levels through simple keywords that modify multiple tolerance parameters simultaneously. These range from SloppySCF for preliminary investigations to ExtremeSCF for maximum precision approaching numerical limits [18]. The TightSCF preset is particularly recommended for transition metal complexes, setting energy change tolerance (TolE) to 1e-8, RMS density change (TolRMSP) to 5e-9, and maximum density change (TolMaxP) to 1e-7 [18].
Q-Chem utilizes the SCF_CONVERGENCE variable with integer values corresponding to 10^(-n) thresholds, defaulting to 5 for single-point energies and 7 for geometry optimizations and frequency calculations [10] [59]. The software automatically adjusts integral thresholds (THRESH) to maintain compatibility with the requested SCF convergence criteria.
ADF's specific convergence tolerance framework was not detailed in the available search results, though as a mature DFT-focused package it undoubtedly provides customizable convergence controls.
Table 2: Standard SCF Convergence Tolerance Presets
| Tolerance Level | ORCA (TolE) | Q-Chem (SCF_CONVERGENCE) | Typical Application Context |
|---|---|---|---|
| Preliminary | 3e-5 (Sloppy) |
4 | Initial geometry scans, large systems |
| Default | 1e-6 (Medium) |
5-6 | Standard single-point energies |
| Enhanced | 1e-8 (Tight) |
7 | Transition metal complexes, property calculations |
| Ultra-Precise | 1e-9 (VeryTight) |
8 | Frequency calculations, sensitive properties |
Experimental Protocols for Pathological Convergence Cases
Protocol 1: Open-Shell Transition Metal Complexes
Background: Open-shell transition metal compounds represent one of the most challenging cases for SCF convergence due to dense electronic states, near-degeneracies, and complex potential energy surfaces. The protocol below outlines a systematic approach for achieving convergence in these systems.
Materials and Software:
- Quantum chemistry software (ORCA, Q-Chem, or ADF)
- Molecular coordinate file of the target complex
- High-performance computing resources
- Visualization software (e.g., Chemcraft [60]) for orbital analysis
Methodology:
Step 1: Initial System Preparation
- Begin with a reasonable molecular geometry, ideally from crystallographic data or a pre-optimized structure using semi-empirical methods
- Verify molecular charge and multiplicity appropriate for the metal center and ligands
- For ORCA: Prepare input file with coordinate block specifying charge and multiplicity
Step 2: Preliminary Calculation with Conservative Settings
- Employ a moderate-sized basis set without diffuse functions initially (e.g., def2-SVP for ORCA, 6-31G* for Q-Chem)
- Use stable functional (e.g., BP86 or B3LYP) before advancing to more complex functionals
- Implement moderate convergence criteria (e.g.,
MediumSCFin ORCA orSCF_CONVERGENCE 5in Q-Chem)
Step 3: Specialized Algorithm Selection ORCA Implementation:
Q-Chem Implementation:
Step 4: Advanced Techniques for Persistent Cases
- If standard approaches fail, employ level shifting (ORCA:
%scf Shift 0.1, ErrOff 0.1 end) - For ORCA: Disable TRAH if it slows convergence excessively using
! NoTrah - For Q-Chem: Implement stability analysis via
STABILITY_ANALYSIS trueto check for wavefunction instabilities - Utilize
SCF_GUESS_MIXin Q-Chem to break alpha-beta symmetry in stubborn open-shell cases
Step 5: Progressive Basis Set and Functional Refinement
- Once converged with moderate settings, systematically increase basis set quality
- Gradually introduce diffuse functions if needed for property calculations
- Transition to more sophisticated functionals only after establishing stable convergence
Validation and Troubleshooting:
- Confirm convergence by examining both energy change and density change criteria
- Use visualization tools like Chemcraft to examine molecular orbitals and identify problematic orbital interactions [60]
- For oscillating solutions, increase damping parameters or implement more aggressive DIIS subspace management
Protocol 2: Conjugated Radical Anions with Diffuse Functions
Background: Conjugated radical anions with diffuse basis functions present exceptional convergence challenges due to weakly bound electrons and near-linear dependencies in the basis set. This protocol addresses these specific pathological characteristics.
Materials and Software:
- Quantum chemistry software with robust handling of diffuse functions (ORCA recommended)
- Appropriate diffuse basis sets (e.g., aug-cc-pVDZ, ma-def2-TZVP)
- Computational resources with sufficient memory for handling integral evaluation
Methodology:
Step 1: Basis Set Selection and Linear Dependence Management
- Select appropriate diffuse basis sets but be prepared to address linear dependencies
- For ORCA: Use
%scf DirectResetFreq 1 endto reduce numerical noise by frequent Fock matrix rebuilds - Consider automatically removing linear dependencies via software-specific options
Step 2: Initial Guess Generation
- Converge a simpler calculation without diffuse functions first
- For ORCA: Read orbitals from previous calculation using
! MOReadand%moinp "previous.gbw" - Alternatively, try converging a 1- or 2-electron oxidized closed-shell state, then use those orbitals as a starting point
Step 3: Specialized SCF Configuration ORCA-Specific Implementation:
Step 4: Convergence Monitoring and Adjustment
- Carefully monitor initial SCF iterations for oscillatory behavior
- If oscillations occur, implement additional damping or reduce the DIIS subspace size
- For ORCA: If TRAH activates but progresses slowly, adjust AutoTRAH parameters:
Step 5: Progressive Methodology
- Begin with Hartree-Fock before advancing to DFT, as HF sometimes converges more readily
- Employ range-separated functionals if standard functionals fail
- Consider double-hybrid functionals only after establishing robust convergence
Validation:
- Verify reasonable orbital energies and occupations
- Confirm expectation values match chemical intuition (e.g., spin densities, population analysis)
- Check for absence of intruder states or anomalous virtual orbital occupations
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Critical Computational Reagents for SCF Convergence Research
| Reagent/Solution | Function | Implementation Examples |
|---|---|---|
| Algorithmic Dampers | Controls large oscillations in early SCF iterations | ORCA: SlowConv, VerySlowConvQ-Chem: RCA algorithm |
| Second-Order Convergers | Provides quadratic convergence near solution | ORCA: TRAH, SOSCFQ-Chem: GDM |
| DIIS Subspace Management | Balances convergence stability and memory usage | ORCA: DIISMaxEqQ-Chem: DIIS_SUBSPACE_SIZE |
| Level Shifters | Stabilizes convergence by shifting virtual orbitals | ORCA: %scf Shift 0.1 endQ-Chem: LEVEL_SHIFT |
| Initial Guess Handlers | Provides alternative starting orbitals | ORCA: PAtom, Hueckel, HCore guessesQ-Chem: SCF_GUESS_MIX |
| Integral Direct Methods | Reduces numerical noise in Fock builds | ORCA: DirectResetFreqQ-Chem: DIRECT_SCF |
Decision Framework and Workflow Integration
The selection of an appropriate software package and configuration strategy depends on multiple factors including system characteristics, available computational resources, and researcher expertise. The following decision framework provides guidance for selecting optimal approaches.
Software Selection Guidelines
Based on the comparative analysis, the following software-specific recommendations emerge:
ORCA demonstrates particular strength for:
- Open-shell transition metal complexes utilizing its specialized
SlowConvand SOSCF algorithms - Systems requiring automatic algorithm switching via the TRAH protocol
- Cases benefiting from extensive specialized keywords developed specifically for pathological convergence
- Researchers preferring predefined convergence presets with option for granular control
Q-Chem offers advantages for:
- Cases where DIIS demonstrates initial progress but fails to fully converge (using DIIS_GDM algorithm)
- Restricted open-shell systems where GDM provides superior performance
- Situations requiring wavefunction stability analysis
- Researchers desiring explicit algorithmic control and modular SCF components
ADF, while less extensively documented in the available sources for SCF convergence specifics, provides robust density functional capabilities particularly suited for:
- Transition metal containing systems
- Materials with heavy elements
- Researchers focused specifically on DFT methodologies
This comparative analysis demonstrates that while ORCA, Q-Chem, and ADF share the common goal of achieving SCF convergence, their approaches, algorithmic implementations, and specialization areas differ significantly. ORCA provides extensive automated and specialized tools for the most challenging cases, particularly open-shell transition metal systems. Q-Chem offers robust fallback algorithms and modular control, with exceptional capabilities in geometric direct minimization. ADF brings strong DFT-specific implementations suited for metal-containing systems and materials.
For researchers pursuing pathological convergence cases, ORCA's specialized keywords and automated algorithm switching provide a powerful first line of attack, particularly when supplemented by Q-Chem's GDM algorithm for particularly stubborn cases. The protocols and decision framework presented herein offer practical pathways for addressing even the most challenging convergence scenarios, providing computational chemists and drug development researchers with structured methodologies for extending the range of tractable systems in their research.
Future directions in this field will likely include increased integration of machine learning approaches for initial guess generation [61], enhanced automatic algorithm selection based on system characteristics, and continued refinement of second-order methods for improved computational efficiency. The ongoing development of large language models for quantum chemistry input generation [61] may also democratize access to advanced SCF convergence techniques, allowing non-specialists to implement sophisticated protocols through natural language interfaces.
Best Practices for Documentation and Reproducibility in SCF Studies
Self-Consistent Field (SCF) methods are fundamental to computational chemistry, enabling the calculation of molecular electronic structure. However, pathological convergence cases—often encountered with open-shell transition metal complexes, systems with multireference character, or at extreme geometries—pose significant challenges to obtaining physically meaningful results. Achieving reproducibility in these demanding calculations requires meticulous documentation and a systematic approach to SCF protocol application. This document provides detailed application notes and experimental protocols, framed within a broader thesis on SCF block settings, to guide researchers in overcoming non-convergence and ensuring their work is robust, reliable, and reproducible.
The SCF procedure is an iterative algorithm that searches for a self-consistent electron density. Convergence is typically assessed based on changes in energy and the density matrix between cycles [9]. Pathological systems violate the underlying assumptions of standard SCF methods, leading to oscillations, stalling, or divergence. Common characteristics include:
- Open-shell electronic configurations, particularly in transition metal compounds [1].
- Near-degenerate orbitals leading to small HOMO-LUMO gaps.
- Geometries far from equilibrium, where the initial density guess is poor and the electronic structure may be multireference in nature [5].
Standard convergence accelerators like DIIS (Direct Inversion in the Iterative Subspace) can fail for these systems, necessitating a structured troubleshooting workflow and comprehensive reporting.
Troubleshooting Protocol for Pathological SCF Convergence
The following step-by-step protocol is designed to systematically address SCF non-convergence. The corresponding workflow is illustrated in Figure 1.
Preliminary Checks and Initial Stabilization
Objective: To eliminate simple causes of failure and stabilize the initial SCF iterations.
- Geometry Inspection: Visually inspect the molecular geometry. Pathological geometries, such as atoms at extreme separations, are a common root cause [5]. If the geometry is unreasonable, consider if it is physically relevant to your study.
- Increase Maximum Iterations: If the SCF is slowly converging but runs out of cycles, simply increase the maximum iteration count (e.g., to 500) [1].
- ORCA Input:
%scf MaxIter 500 end - BAND Input: In the
SCFblock, setIterations 500[9].
- ORCA Input:
- Apply Damping: For wild oscillations in the initial SCF iterations, employ damping algorithms.
Advanced Algorithm Switching
Objective: To employ more robust, but computationally expensive, SCF solvers.
- Activate Second-Order Convergers: If damping and DIIS fail, switch to a second-order convergence algorithm.
- SOSCF Fine-Tuning: The SOSCF algorithm can itself fail with "huge, unreliable step" errors. If this occurs, disable it with
!NOSOSCFor delay its startup by lowering the orbital gradient threshold [1].- ORCA Input:
Manipulation of the Initial Guess and Electronic State
Objective: To provide a better starting point for the SCF procedure.
- Alternative Initial Guesses: Switch from the default
PModelguess toPAtom,Hueckel, orHCore[1]. - Converge a Simpler System:
- Method/ Basis Set Downgrade: Converge the SCF using a lower level of theory (e.g., HF or BP86) and a smaller basis set (e.g., def2-SVP). Use the resulting orbitals as a guess for the target calculation via
! MORead[1]. - Converge a Different Charge/Spin State: Attempt to converge a closed-shell cation or a high-spin multiplet (e.g., a septet) of the system. The orbitals from this calculation can sometimes serve as a productive guess for the target electronic state [5].
- Method/ Basis Set Downgrade: Converge the SCF using a lower level of theory (e.g., HF or BP86) and a smaller basis set (e.g., def2-SVP). Use the resulting orbitals as a guess for the target calculation via
- Use a Core-Hole or Ionized Guess: For systems with severe convergence issues, converging a 1- or 2-electron oxidized state and using its orbitals as a guess can be effective [1].
Final Resort Strategies for Intractable Cases
Objective: To force convergence in truly pathological systems, accepting a significant increase in computational cost.
- Aggressive DIIS Settings: Increase the memory and reset frequency of the DIIS algorithm.
- ORCA Input:
- Orbital Smearing: Introduce a finite electronic temperature to smear orbital occupations around the Fermi level, breaking degeneracies that hinder convergence [9].
- BAND Input: The
Degeneratekey in theConvergenceblock can be used for this purpose [9].
- BAND Input: The
Figure 1: Systematic Workflow for Troubleshooting SCF Convergence
Quantitative Data Presentation and Analysis
Effective documentation relies on the clear presentation of quantitative data related to SCF performance. The tables below provide templates for reporting convergence criteria and algorithmic settings.
Table 1: Default SCF Convergence Criteria in the BAND Code (Adapted from [9])
| NumericalQuality Setting | Convergence Criterion (Formula) | Description |
|---|---|---|
| Basic | 1e-5 × √(N_atoms) | Least stringent, faster calculations |
| Normal | 1e-6 × √(N_atoms) | Default setting for balanced accuracy |
| Good | 1e-7 × √(N_atoms) | Higher accuracy requirement |
| VeryGood | 1e-8 × √(N_atoms) | Most stringent, for high-precision work |
Table 2: Advanced SCF Algorithm Settings for Pathological Cases in ORCA [1]
| SCF Block Keyword | Default Value | Recommended Value for Pathological Cases | Function |
|---|---|---|---|
MaxIter |
125 | 500 - 1500 | Maximum number of SCF cycles allowed |
DIISMaxEq |
5 | 15 - 40 | Number of Fock matrices in DIIS extrapolation |
directresetfreq |
15 | 1 | Frequency of full Fock matrix rebuild; 1 = every cycle |
SOSCFStart |
0.0033 | 0.00033 | Orbital gradient threshold to start SOSCF |
Reproducibility Framework and Documentation Standards
Reproducibility requires that any researcher can exactly replicate the computational environment and procedure. Adopting standards from initiatives like the SC Conference Reproducibility Initiative is critical [62].
Mandatory Documentation Protocol
The following items must be documented for every publication involving non-trivial SCF calculations:
- Software and Versioning: The computational chemistry package (e.g., ORCA 5.0.3, PSI4 1.8) must be explicitly stated, including the build date if possible.
- Complete Input File: The entire input file, including all molecular coordinates, molecular charge, spin multiplicity, and convergence criteria, must be provided in the supplementary information.
- SCF Algorithm and Settings: The specific SCF method (DIIS, TRAH, SOSCF, KDIIS) and all non-default parameters (e.g.,
%scfblock in ORCA,Convergenceblock in BAND) must be reported. This includes the final convergence threshold. - Initial Guess Strategy: The method used for the initial orbital guess (e.g.,
PModel,HCore, orMOReadfrom a previous calculation) must be declared. - Basis Set and Hamiltonian: The full name of the basis set and any effective core potentials, or the DFT functional and grid, must be specified.
Artifact Description and Evaluation
Following modern reproducibility standards, authors should provide:
- An Artifact Description (AD) Appendix: A mandatory description of the computational artifacts (input files, scripts, etc.) [62].
- An Artifact Evaluation (AE) Appendix: An optional, but highly encouraged, appendix that provides workflows (e.g., shell scripts, container images) that allow reviewers to automatically reproduce key results [62]. Achieving ACM "Available," "Functional," and "Reproducible" badges is a mark of high-quality computational research.
The Scientist's Toolkit: Research Reagent Solutions
This table details the essential "reagents" or computational tools required for advanced SCF studies.
Table 3: Essential Research Reagent Solutions for SCF Studies
| Item Name | Function/Brief Explanation | Example Use Case |
|---|---|---|
| DIIS (Direct Inversion in Iterative Subspace) | Extrapolates Fock matrices from previous cycles to accelerate convergence. | Standard converger for well-behaved, closed-shell organic molecules. |
| TRAH (Trust Radius Augmented Hessian) | A robust second-order SCF converger that is more stable but slower than DIIS. | Automatically activated in ORCA when DIIS fails; ideal for open-shell transition metal complexes [1]. |
| SOSCF (Second-Order SCF) | Uses the exact Hessian to converge the SCF once the orbital gradient is small. | Speeds up final convergence after initial stabilization via damping; can be unstable for some open-shell systems [1]. |
| Orbital Smearing (Fermi Smearing) | Assigns fractional occupations to orbitals near the Fermi level, breaking degeneracies. | Essential for converging metallic systems or calculations with small HOMO-LUMO gaps [9]. |
| MORead (Orbital Reading) | Uses orbitals from a previous calculation as the initial guess for a new SCF procedure. | Critical protocol for generating an initial guess from a converged, simpler calculation (e.g., BP86) for a more complex one (e.g., hybrid DFT) [1]. |
Conclusion
Successfully managing pathological SCF convergence requires a multifaceted strategy that combines deep theoretical understanding with practical algorithmic adjustments. This guide has synthesized key approaches: identifying the root causes of failure, implementing advanced second-order convergence algorithms, applying systematic troubleshooting protocols, and rigorously validating the physical meaningfulness of obtained solutions. For biomedical researchers, mastering these techniques is particularly crucial for accurately modeling challenging systems like metalloenzymes or reactive drug intermediates. Future directions will likely involve increased automation in SCF convergence, machine learning-assisted initial guesses, and enhanced algorithms specifically designed for high-throughput screening in drug discovery. By adopting these robust convergence strategies, computational chemists can significantly improve the reliability of their calculations, leading to more confident predictions in drug development pipelines.
References
- 1. SCF Convergence Issues - ORCA Input Library [https://sites.google.com/site/orcainputlibrary/scf-convergence-issues]
- 2. Common Errors in Density-Functional Theory - Rowan Scientific [https://rowansci.com/blog/dft-errors]
- 3. What are the physical reasons if the SCF doesn't converge? [https://mattermodeling.stackexchange.com/questions/8635/what-are-the-physical-reasons-if-the-scf-doesnt-converge]
- 4. Difficult cases for converging Kohn-Sham SCF calculations [https://mattermodeling.stackexchange.com/questions/495/difficult-cases-for-converging-kohn-sham-scf-calculations]
- 5. SCF convergence at pathological geometries [https://forum.psicode.org/t/scf-convergence-at-pathological-geometries/326]
- 6. Projects: Organization, Sharing, and Saving Structures [https://rowansci.substack.com/p/projects-organization-sharing-and]
- 7. 7.9. DeltaSCF: Converging to Arbitrary Single-Reference ... [https://www.faccts.de/docs/orca/6.0/manual/contents/detailed/deltascf.html]
- 8. Variation from closed-shell to open shell electronic structures in oligothiophene bis(dioxolene) complexes - Chemical Science (RSC Publishing) DOI:10.1039/D3SC02341A [https://pubs.rsc.org/en/content/articlehtml/2023/sc/d3sc02341a]
- 9. Self Consistent Field (SCF) — BAND 2025.1 documentation [https://www.scm.com/doc/BAND/Accuracy_and_Efficiency/SCF.html]
- 10. 4.5 Converging SCF Calculations [https://manual.q-chem.com/5.1/sect-convergence.html]
- 11. Self-consistent field (SCF) methods [https://pyscf.org/user/scf.html]
- 12. Why do I see Oscillations in energy using SCF Hartree ... [https://physics.stackexchange.com/questions/354404/why-do-i-see-oscillations-in-energy-using-scf-hartree-fock-approximation]
- 13. SCF Convergence Guidelines for ADF - SCM [https://www.scm.com/doc/ADF/Rec_problems_questions/SCF.html]
- 14. How can you manage SCF convergence problems? [https://chemistry.stackexchange.com/questions/27008/how-can-you-manage-scf-convergence-problems]
- 15. Assessment of Initial for Self-Consistent Field Guesses . Calculations [https://pubmed.ncbi.nlm.nih.gov/30653322/]
- 16. 4.4 SCF Initial Guess [https://manual.q-chem.com/4.3/sect-initialguess.html]
- 17. 7.6. Choice of Initial Guess and Restart of SCF Calculations [https://www.faccts.de/docs/orca/6.0/manual/contents/detailed/initguess.html]
- 18. 7.7. SCF Convergence - ORCA 6.0 Manual [https://www.faccts.de/docs/orca/6.0/manual/contents/detailed/scfconv.html]
- 19. Functional, Orbital, and Eigenvalue-Driven Analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11948335/]
- 20. 2.6. Self-Consistent-Field (SCF) [https://orca-manual.mpi-muelheim.mpg.de/contents/essentialelements/scf.html]
- 21. Numerical precision - ORCA Input Library [https://sites.google.com/site/orcainputlibrary/numerical-precision]
- 22. 4.5.9 Relaxed Constraint Algorithm (RCA) [https://manual.q-chem.com/5.4/Ch4.S5.SS9.html]
- 23. 4.5.2 Basic Convergence Control Options [https://manual.q-chem.com/6.3/Ch4.S5.SS2.html]
- 24. 4.5.7 Geometric Direct Minimization (GDM) [https://manual.q-chem.com/5.3/sect_gdm.html]
- 25. 4.5 Converging SCF Calculations [https://manual.q-chem.com/4.3/sect-convergence.html]
- 26. 4.5.12 User-Customized Hybrid SCF Algorithm [https://manual.q-chem.com/6.3/sub_custom_algorithm.html]
- 27. 6.14.6 Convergence Strategies and More Advanced Options [https://manual.q-chem.com/latest/sect_activeopt.html]
- 28. SCF — ADF 2025.1 documentation - SCM [https://www.scm.com/doc/ADF/Input/SCF.html]
- 29. Accelerating self-consistent field convergence with the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2830258/]
- 30. Electronic Configuration — ADF 2025.1 documentation - SCM [https://www.scm.com/doc/ADF/Input/Electronic_Configuration.html]
- 31. Three-temperature model for predicting critical ... [https://www.nature.com/articles/s41524-025-01798-w]
- 32. Surface-Induced Electronic and Vibrational Level Shifting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10532516/]
- 33. Surface-Induced Electronic and Vibrational Level Shifting ... [https://www.mdpi.com/1996-1944/16/18/6150]
- 34. 4.5.3 Direct Inversion in the Iterative Subspace (DIIS) [https://manual.q-chem.com/5.2/Ch4.S5.SS3.html]
- 35. 4.5.4 Damping‣ 4.5 Converging SCF Calculations ‣ ... [https://manual.q-chem.com/5.3/sect_damp.html]
- 36. A Reusable Library for Second-Order Orbital Optimization ... [https://arxiv.org/html/2509.13931v1]
- 37. Accelerating ab initio molecular dynamics simulations by ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261416306170]
- 38. Rust and redemption: iron–sulfur clusters and oxygen in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12241848/]
- 39. Mechanisms Operating in the Use of Transition Metal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12298408/]
- 40. Iron–Sulfur Clusters: Assembly and Biological Roles [https://www.mdpi.com/2304-6740/12/8/216]
- 41. A new therapeutic perspective on metal-based drugs [https://pubs.rsc.org/en/content/articlehtml/2025/dt/d5dt01567g]
- 42. Iron-sulfur cluster assembly [https://bio-protocol.org/exchange/minidetail?id=7531007&type=30]
- 43. Iron-Sulfur Cluster Engineering Provides Insight into the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3288176/]
- 44. Teaching oxidation states to neural networks [https://www.nature.com/articles/s41524-025-01709-z]
- 45. The Conundrum of Diffuse Basis Sets: A Blessing for ... [https://arxiv.org/html/2502.04631v1]
- 46. SCF [https://psicode.org/psi4manual/master/autodir_options_c/module__scf.html]
- 47. Basis Set Effect on Linear Response Density Functional ... [https://chemrxiv.org/engage/chemrxiv/article-details/68e3ee3c5dd091524f1daa6f]
- 48. The importance of diffuse functions in basis sets to produce ... [https://www.sciencedirect.com/science/article/abs/pii/S000926141930226X]
- 49. Excited state dipole moments from ΔSCF: a benchmark [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp01695a]
- 50. 7.11. SCF Stability Analysis - ORCA 6.0 Manual [https://www.faccts.de/docs/orca/6.0/manual/contents/detailed/stabilityanalysis.html]
- 51. 4.5.15 Internal Stability Analysis and Automated Correction ... [https://manual.q-chem.com/5.4/sub_stability_analysis.html]
- 52. 2.16. SCF Stability Analysis [https://orca-manual.mpi-muelheim.mpg.de/contents/essentialelements/stabilityanalysis.html]
- 53. Spin Contamination [https://server.ccl.net/cca/documents/dyoung/topics-orig/spin_cont.html]
- 54. Dealing with Spin Contamination - Dr. Joaquin Barroso's Blog [https://joaquinbarroso.com/2017/06/20/dealing-with-spin-contamination/]
- 55. Performances of recently-proposed functionals for ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261410015435]
- 56. Performance of quantum chemistry methods for a ... [https://www.sciencedirect.com/org/science/article/pii/S2041652024018029]
- 57. Benchmarking Density Functionals for Ground- and Excited ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12257512/]
- 58. GAMESS, Gaussian - software for Quantum Chemistry [https://www.biomolecular-modeling.com/Software_Quantum_Chemistry.html]
- 59. 4.3 Basic SCF Job Control [https://manual.q-chem.com/5.0/sec-SCF_job-control.html]
- 60. Description [https://www.chemcraftprog.com/description.html]
- 61. Developing large language models for quantum chemistry ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00366g]
- 62. Reproducibility Initiative - SC25 - SC Conference [https://sc25.supercomputing.org/program/papers/reproducibility-initiative/]