Basin-Hopping Method: A Complete Guide to Global Optimization for Drug Discovery and Biomolecular Structure Prediction
This article provides a comprehensive exploration of the Basin-Hopping (BH) algorithm, a powerful global optimization technique essential for navigating complex potential energy surfaces in computational chemistry and drug discovery.
Basin-Hopping Method: A Complete Guide to Global Optimization for Drug Discovery and Biomolecular Structure Prediction
Abstract
This article provides a comprehensive exploration of the Basin-Hopping (BH) algorithm, a powerful global optimization technique essential for navigating complex potential energy surfaces in computational chemistry and drug discovery. Tailored for researchers and drug development professionals, the content covers foundational principles, practical implementation strategies, and advanced optimization techniques. It details BH applications in identifying low-energy molecular conformations and atomic cluster structures, addresses common challenges and solutions for efficient searching, and offers a comparative analysis with other metaheuristics. The guide also synthesizes key takeaways and discusses future directions, highlighting BH's transformative potential in biomedical research for accelerating lead compound optimization and improving predictive accuracy in structure-based drug design.
Understanding Basin-Hopping: Core Principles and the Challenge of Rugged Energy Landscapes
Defining the Global Optimization Problem in Chemical Systems
For researchers in computational chemistry and drug development, the global optimization problem represents a fundamental challenge: finding the most stable configuration of a chemical system among a vast number of possibilities. This problem requires locating the global minimum on a complex, multidimensional potential energy surface (PES), which represents the most thermodynamically stable structure of a molecule or molecular assembly [1]. The complexity arises because the number of local minima on these surfaces grows exponentially with system size, following a relation of the form (N_{\text{min}}(N) = \exp(\xi N)), where (N) is the number of atoms and (\xi) is a system-dependent constant [1].
Within this context, the basin-hopping algorithm has emerged as a powerful stochastic global optimization method that combines random perturbation of coordinates with local minimization to efficiently explore complex energy landscapes [2] [3]. This method has proven particularly valuable for predicting molecular conformations, protein structures, crystal polymorphs, and reaction pathways—all critical tasks in computational chemistry and drug design [1].
Frequently Asked Questions (FAQs)
What defines a "global minimum" in chemical systems? The global minimum is the molecular geometry corresponding to the lowest point on the potential energy surface (PES). It represents the most thermodynamically stable structure of a molecule or molecular cluster, which is essential for accurately predicting properties including thermodynamic stability, reactivity, spectroscopic behavior, and biological activity [1].
How does basin-hopping differ from simulated annealing? While both are stochastic global optimization methods, basin-hopping incorporates a local minimization step after each random perturbation, effectively transforming the PES into a collection of interpenetrating staircases. This allows the algorithm to "hop" between local minima more efficiently than simulated annealing, particularly for "funnel-like, but rugged" energy landscapes [3] [4].
Can basin-hopping guarantee finding the true global minimum? No. Basin-hopping is not a deterministic method, and identifying the true global minimum with absolute certainty in a finite, stochastic search is not guaranteed. Confidence in results typically comes from satisfactory agreement with experimental observations and consistency across multiple parallel simulations with different initial conditions [4].
What are the most critical parameters to adjust in a basin-hopping simulation? Two crucial parameters are the step size for random displacements and the temperature
Tused in the Metropolis acceptance criterion. The step size should be comparable to the typical separation between local minima, whileTshould be comparable to the typical energy difference between local minima for optimal performance [3].How can I improve the efficiency of my basin-hopping calculations? Recent research has shown that augmenting basin-hopping with unsupervised machine learning techniques, such as similarity indices and hierarchical clustering, can significantly improve efficiency by better tracking explored regions of the PES and guiding searches toward unexplored areas [4].
Troubleshooting Common Experimental Issues
Problem 1: Algorithm Becoming Trapped in Local Minima
Symptoms: The optimization consistently converges to the same high-energy structure across multiple runs, failing to locate lower-energy minima.
Solutions:
- Increase temperature parameter: Raise the "temperature"
Tin the Metropolis criterion to allow acceptance of more uphill moves, facilitating escape from deep local minima [3]. - Adjust step size: Increase the maximum step size for random displacements to encourage exploration of wider regions of the configuration space [3].
- Implement enhanced sampling: Combine with parallel tempering or use adaptive step-size adjustment based on target acceptance rates [3] [5].
- Utilize machine learning augmentation: Apply similarity indices and hierarchical clustering to identify unexplored regions of the PES and direct searches accordingly [4].
Problem 2: Poor Computational Efficiency with Large Systems
Symptoms: Calculation times become prohibitively long as system size increases, with slow convergence.
Solutions:
- Optimize local minimizer: Choose efficient local minimization methods (e.g., L-BFGS-B) and leverage gradient information where available [3].
- Selective degree of freedom manipulation: Freeze chemically bonded distances and focus random perturbations on specific degrees of freedom such as dihedral angles [4].
- Implement confidence-based dimensionality reduction: For high-dimensional problems, employ rule-based sensor confidence evaluation to reduce the number of parameters requiring optimization [5].
Problem 3: Inaccurate Energy Evaluations
Symptoms: Located minima do not correspond to experimentally observed structures, despite extensive sampling.
Solutions:
- Improve potential energy function: Refine force field parameters or use higher-level theoretical methods (e.g., DFT instead of molecular mechanics) for more accurate energy evaluations [4].
- Incorporate bioinformatic data: Use associative memory Hamiltonians that flexibly incorporate bioinformatic data to improve structure prediction potentials [2].
- Validate with multiple methods: Cross-check putative global minima using different optimization algorithms or initial conditions [6].
Experimental Protocols & Workflows
Standard Basin-Hopping Implementation
The core basin-hopping algorithm follows an iterative cycle with these steps [3] [4]:
- Random Perturbation: Apply random changes to the current molecular coordinates
- Local Minimization: Perform local energy minimization starting from the perturbed geometry
- Accept/Reject Decision: Apply Metropolis criterion based on the minimized energy difference from the current structure
- Update: If accepted, update the current structure; otherwise, retain the previous structure
- Iterate: Repeat for a specified number of iterations or until convergence criteria are met
Enhanced BH with Machine Learning Guidance
For complex systems, augment basin-hopping with unsupervised machine learning techniques [4]:
- Perform initial BH sampling to generate diverse structures
- Calculate similarity indices between all identified local minima
- Apply hierarchical clustering to group similar structures
- Identify undersampled regions of the conformational space
- Guide subsequent BH steps toward unexplored regions
- Iterate until conformational space is adequately mapped
Comparative Methodologies
Table 1: Classification of Global Optimization Methods in Chemical Systems
| Method Type | Examples | Key Characteristics | Best Use Cases |
|---|---|---|---|
| Stochastic | Basin-Hopping [1] [3], Genetic Algorithms [1], Simulated Annealing [1] | Incorporate randomness; no convergence guarantee; good for complex landscapes | Complex, high-dimensional systems with many local minima |
| Deterministic | Branch-and-Bound [7], Single-Ended Methods [1], Molecular Dynamics [1] | Follow defined rules; use gradient information; precise convergence | Smaller systems where exhaustive search is feasible |
| Hybrid | ML-Augmented BH [4], Uncertainty-Inclusive BH [5] | Combine strengths of multiple approaches; balance exploration/exploitation | Challenging systems requiring both breadth and depth of search |
Table 2: Basin-Hopping Variants and Their Applications
| Variant | Key Features | Reported Applications |
|---|---|---|
| Significant Structures BH (SSBH) [6] | Updates geometry to local minimum after accepted step | Atomic/molecular clusters (Lennard-Jones, benzene) |
| Raw Structures BH (RSBH) [6] | Keeps raw geometry (not minimized) for next step | Less efficient than SSBH in cluster optimization |
| Mutational BH [4] | Incorporates biomolecular-specific moves | Biomolecular structure prediction |
| Uncertainty-Inclusive BH (Un-BH) [5] | Addresses initial point selection uncertainty | High-dimensional sensor calibration in complex systems |
Table 3: Key Software Tools for Global Optimization in Chemical Systems
| Tool Name | Capabilities | Application Context |
|---|---|---|
| GMIN [3] | Fortran implementation of basin-hopping | Atomic clusters, biomolecules |
| SciPy basinhopping [3] | Python implementation with customizable steps | General purpose chemical optimization |
| BARON [7] | Branch-and-reduce optimization | Process design, molecular design, metabolic systems |
| GRRM [1] | Global reaction route mapping | Reaction pathway exploration |
| OGOLEM [8] | Genetic algorithm framework | Crystal structure prediction, clusters |
Advanced Applications in Drug Development
Basin-hopping methodology has demonstrated particular value in pharmaceutical research through these applications:
Protein Structure Prediction: The algorithm has successfully identified low-lying minima for associative memory Hamiltonian structure prediction potentials, locating conformations closer to experimental structures than molecular dynamics with simulated annealing for small systems [2].
Spectroscopic Assignment: BH with machine learning augmentation has been used to assign infrared multiple photon dissociation spectra of protonated serine dimer and other biologically relevant molecules [4].
Ion Mobility Modeling: The method enables determination of temperature-dependent collision cross-sections for flexible peptides like protonated alanine tripeptide, supporting interpretation of ion mobility experiments [4].
For researchers implementing these methods, successful application requires careful parameter tuning, method validation against known systems, and consideration of hybrid approaches that combine basin-hopping with complementary optimization strategies to address the complex global optimization challenges in chemical systems and drug development.
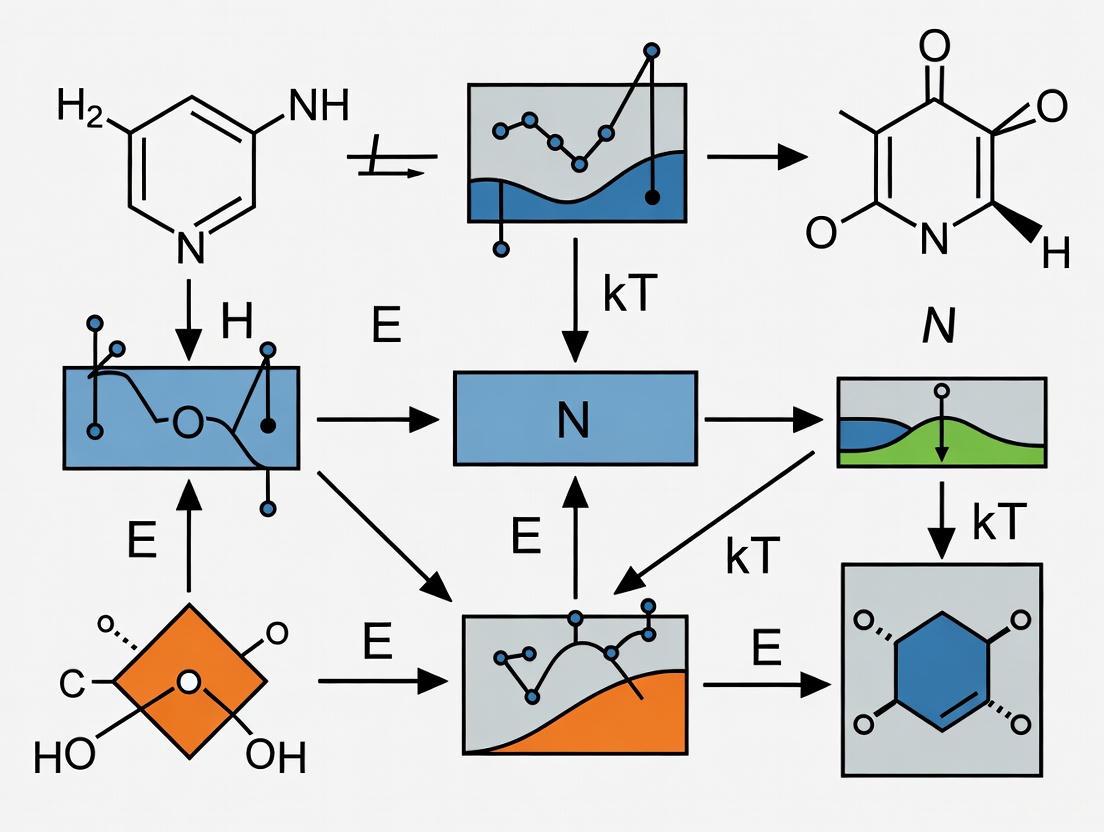
The Basin-Hopping (BH) algorithm is a powerful global optimization technique designed to find the global minimum of a function by navigating complex, multi-modal potential energy surfaces (PES). It is particularly effective for problems in computational chemistry and physics, such as identifying low-energy structures of atomic clusters, molecules, and proteins [9] [2] [4]. The algorithm transforms the original PES into a collection of interpenetrating staircases, where each stair corresponds to the potential energy of a local minimum. This transformation allows the search to efficiently hop between different basins of attraction [4].
The core principle of Basin-Hopping involves a two-phase iterative cycle that combines global exploration with local refinement [10]. The key steps are:
- Random Perturbation: The current coordinates are randomly displaced.
- Local Minimization: The perturbed coordinates are used as a starting point for a local optimization, mapping the system to a local minimum on the PES.
- Acceptance/Rejection: The new local minimum is accepted or rejected based on a probabilistic criterion, often the Metropolis condition used in Monte Carlo algorithms [10].
This process allows the algorithm to escape local minima and explore diverse regions of the energy landscape.
Algorithm Workflow and Methodology
The following diagram illustrates the iterative procedure of the Basin-Hopping algorithm:
Key Steps in the Workflow:
- Initialization: The algorithm begins with an initial guess of the coordinates.
- Perturbation: A random structural displacement is applied to the current coordinates. The magnitude of this step is controlled by the
stepsizeparameter, which is crucial for sampling efficiency [10]. - Local Minimization: The perturbed structure undergoes local energy minimization using methods like L-BFGS-B or BFGS [9] [11]. This step finds the nearest local minimum on the PES.
- Metropolis Criterion: The new minimum is accepted if its energy is lower than the current one. If the energy is higher, it may still be accepted with a probability of
exp( -(E_new - E_old) / T ), whereTis a "temperature" parameter. This stochastic acceptance allows the algorithm to escape local minima [10]. - Iteration and Termination: Steps 2-4 are repeated for a specified number of iterations (
niter) until a stopping condition is met, such as a fixed number of iterations or no improvement in the global minimum candidate for a given number of steps (niter_success) [10].
Key Parameters for Experimental Design
The performance of Basin-Hopping is highly dependent on several key parameters. The table below summarizes these parameters, their functions, and typical configuration values.
| Parameter | Function | Impact on Search | Recommended Value / Range |
|---|---|---|---|
Temperature (T) |
Controls the probability of accepting uphill moves. | Higher T promotes exploration; lower T favors exploitation. |
Should be comparable to the typical function value difference between local minima [10]. |
Step Size (stepsize) |
Controls the maximum displacement during random perturbation. | Too small causes trapping; too large reduces efficiency. | Should be comparable to the typical separation between local minima. Can be set to adapt dynamically [9] [10]. |
Number of Iterations (niter) |
Sets the total number of basin-hopping steps. | More iterations increase the chance of finding the global minimum but require more computation [11]. | Problem-dependent; often requires experimentation (e.g., 100-1000+) [11]. |
| Local Minimizer Method | The algorithm used for local optimization (e.g., BFGS, L-BFGS-B). | Affects the speed and robustness of finding local minima. | L-BFGS-B is common for its efficiency with bounds [9]. |
Troubleshooting Common Experimental Issues
Problem: Algorithm Returns Incorrect Local Minima, Missing Global Minimum This is a fundamental challenge in stochastic global optimization. The algorithm is not guaranteed to find the global minimum in a single run [11] [4].
- Solution 1: Increase Iteration Count. The number of iterations (
niter) may be insufficient. Increasenitersubstantially to allow broader exploration. What works for a small cluster may be entirely inadequate for a complex molecule [11]. - Solution 2: Adjust Temperature (
T). IfTis too low, the algorithm cannot escape deep local minima. IncreaseTto be comparable to the energy barriers between funnels on the PES [10]. - Solution 3: Optimize Step Size (
stepsize). An improperly sized step can hinder exploration. Ifstepsizeis too small, the search is trapped in a subregion; if too large, it becomes a random search. Use an adaptive step size that adjusts to maintain a target acceptance rate (e.g., 0.5) [9] [10]. - Solution 4: Perform Multiple Runs. Execute the algorithm from multiple, diverse starting points (
x0). Consistent results across runs increase confidence that the global minimum has been found [10] [4].
Problem: Prohibitively Long Computation Time for Complex Systems Each local minimization can be costly, especially with high-dimensional systems or expensive energy functions.
- Solution: Implement Parallelization. Instead of evaluating one candidate per step, multiple perturbed candidates can be minimized concurrently. This parallel evaluation significantly reduces wall-clock time and can improve convergence by exploring multiple paths simultaneously [9].
Frequently Asked Questions (FAQs)
Q1: How does Basin-Hopping compare to Molecular Dynamics with Simulated Annealing for protein structure prediction? Basin-Hopping can be more effective at locating lower energy minima and conformations closer to the native experimental structure. This is because its Monte Carlo-minimization approach avoids kinetic traps that can hinder molecular dynamics simulations, allowing for more efficient navigation of the rugged energy landscape [2].
Q2: Can Basin-Hopping be used with electronic structure methods like Density Functional Theory (DFT)? Yes, but the high computational cost of ab initio methods like DFT for each energy and gradient evaluation is a major constraint. A common strategy is to use Basin-Hopping with faster, approximate potentials (e.g., empirical force fields) to find low-energy candidate structures, which are then refined with higher-level DFT calculations [9].
Q3: What are the advanced techniques to improve the efficiency of a Basin-Hopping search? Researchers are augmenting BH with techniques from unsupervised machine learning. This includes:
- Similarity Indices and Clustering: Comparing and clustering found local minima to identify and bias the search towards unexplored regions of the PES.
- Hierarchical Clustering: Building a tree of minima to understand the landscape's funnel topology.
- Dimensionality Reduction: Using methods like multidimensional scaling to visualize the high-dimensional conformation space and guide the search [4].
Q4: Is the global minimum found by Basin-Hopping guaranteed to be the true global minimum? No. Basin-Hopping is a stochastic algorithm, and there is no guarantee that the true global minimum has been found in a finite run. Confidence is built by running the algorithm multiple times from different random starting points and achieving consistent results. Agreement with experimental data also strengthens validation [10] [4].
Essential Research Reagent Solutions
The following table lists key computational "reagents" and their roles in a typical Basin-Hopping experiment for molecular structure search.
| Item | Function in Experiment | Key Consideration |
|---|---|---|
| Potential Energy Function | Calculates the energy and forces for a given atomic configuration. Defines the landscape being searched. | Choice is critical: from fast Lennard-Jones for clusters [9], to molecular mechanics, to accurate but expensive ab initio methods [2]. |
| Local Minimizer (e.g., L-BFGS-B) | Finds the nearest local minimum after each perturbation step [9]. | Must be efficient and robust. The minimizer_kwargs parameter is used to pass settings to this routine [10]. |
Initial Structure (x0) |
The starting configuration for the global search. | Should be diverse if multiple runs are performed. Can be a random configuration or a chemically informed guess [4]. |
| Perturbation Strategy | Generates new trial configurations to escape the current basin of attraction. | The default is random atomic displacements. Custom take_step routines can be defined for problem-specific moves (e.g., dihedral rotations) [10] [4]. |
Mapping Complex Potential Energy Surfaces into Basins of Attraction
FAQs and Troubleshooting Guide
1. My basinhopping calculation is not converging to the global minimum. What should I check? The basin-hopping algorithm's efficiency depends heavily on the choice of its parameters. If you are not finding the global minimum, consider the following:
- Step Size (
stepsize): This is a crucial parameter. The step size for the random displacement should be comparable to the typical separation between local minima in your PES. If it's too small, the algorithm cannot jump to new basins; if it's too large, it becomes inefficient. The algorithm will try to optimize this, but providing a sensible initial value accelerates convergence [10] [12]. - Temperature (
T): The "temperature" parameterTin the Metropolis criterion should be comparable to the typical function value difference between local minima. A higher temperature allows the algorithm to accept uphill moves more readily, helping it escape local minima. IfTis too low, the search may become greedy and get trapped [10]. - Number of Iterations (
niter): For complex, high-dimensional PES, the number of basin-hopping iterations may need to be very large (e.g., thousands) to adequately explore the landscape. You may simply need to run the algorithm for more iterations [12]. - Local Minimizer: Ensure the local minimization algorithm is appropriate for your problem. You can specify this via
minimizer_kwargs. For bounded problems, "L-BFGS-B" is a good default, while "Nelder-Mead" can be used for problems where derivatives are unavailable [12].
2. How can I define a custom acceptance test or step-taking routine? Basin-hopping allows for high customization. You can define your own functions and pass them to the algorithm.
- Custom
accept_test: This function can be used to force the acceptance of a step under specific conditions, for instance, to escape a known problematic local minimum. The function must accept the parametersf_new,x_new,f_old, andx_old, and returnTrue,False, or the string"force accept"[10]. - Custom
take_step: You can replace the default random displacement with a more intelligent perturbation tailored to your system's geometry. This function can optionally have astepsizeattribute that the algorithm will adjust automatically [10].
3. The computation is too slow for my high-dimensional system. Are there alternatives? For very high-dimensional systems, fully mapping the basins of attraction (BoAs) with pointwise methods can be prohibitively expensive due to the "curse of dimensionality" [13]. Recent research focuses on data-efficient methods. One advanced approach is the Boundary-Oriented Progressive Learning Method (BOPLM), which discretizes the state space and uses a machine learning classifier to progressively learn the boundaries between basins, requiring far fewer samples than traditional methods like Monte Carlo simulations [13].
4. What is the significance of a "saddle point" on a Potential Energy Surface? A saddle point, or transition state, is a critical point on the PES that represents the highest energy structure along the lowest energy pathway connecting two minima (e.g., reactants and products). It is a minimum in all directions except one, along which it is a maximum. Identifying saddle points is essential for understanding reaction kinetics and barriers [14] [15].
Basin-Hopping Parameters and Methodologies
Summary of Key Basin-Hopping Parameters
The following table summarizes the core parameters for the scipy.optimize.basinhopping function, which are critical for troubleshooting your experiments [10] [12].
| Parameter | Description | Troubleshooting Tip |
|---|---|---|
niter |
Number of basin-hopping iterations. | Increase for complex landscapes; can require 1000s of iterations [12]. |
T |
"Temperature" for the Metropolis acceptance criterion. | Set comparable to the typical energy difference between local minima [10]. |
stepsize |
Maximum step size for the random displacement. | Should be comparable to the characteristic distance between local minima [10] [12]. |
minimizer_kwargs |
Arguments passed to the local minimizer. | Specify the local method (e.g., {'method':'L-BFGS-B'}) and any required derivatives [10] [12]. |
niter_success |
Stop if the global minimum candidate is unchanged for this many iterations. | Provides an automatic stopping criterion for well-behaved landscapes [10]. |
Detailed Experimental Protocol: Basin-Hopping for Minima Identification This protocol outlines the steps to identify local minima on a PES using the basin-hopping method as implemented in SciPy.
- Define the Objective Function: The function
func(x)must be defined, wherexis the vector of coordinates representing a point on the PES (e.g., atomic positions). The function returns the energy (or other property) at that point [10]. - Choose an Initial Guess (
x0): Select a starting point for the global optimization. - Set Algorithm Parameters:
niter: Based on the problem's complexity, set a sufficiently large number of iterations (e.g., 100 to 10,000).stepsize: Estimate a reasonable step size from known system properties.T: Set the temperature based on known energy barriers or through experimentation.minimizer_kwargs: Define the local minimizer and its options. For example:minimizer_kwargs={"method": "L-BFGS-B", "jac": gradient_function}[10] [12].
- Execute the Algorithm: Run the
basinhoppingfunction with the defined parameters. - Analyze the Result: The returned
OptimizeResultobject contains the coordinates of the found minimum (x), the function value at that minimum (fun), and the full result from the successful local minimization at the lowest minimum (lowest_optimization_result), which can be used for further analysis [10].
The Scientist's Toolkit: Essential Research Reagents & Materials
The following table lists key computational "reagents" and tools used in mapping PES and conducting basin-hopping simulations.
| Item | Function / Explanation |
|---|---|
| Potential Energy Surface (PES) | A function that describes the energy of a system (e.g., a molecule) in terms of its nuclear coordinates. It is the foundational landscape on which optimization occurs [14] [15]. |
| Local Minimization Algorithm (e.g., L-BFGS-B) | The algorithm used at each basin-hopping step to find the local minimum of a basin. It must be efficient and robust [10] [12]. |
| Born-Oppenheimer Approximation | A key approximation that allows the separation of electronic and nuclear motion, making the computation of the PES feasible by assuming nuclei are stationary relative to electrons [15]. |
| Metropolis Criterion | The probabilistic rule used to decide whether to accept or reject a new step based on the change in energy and the algorithm's temperature T. This allows the algorithm to escape local minima [10]. |
| CAMEO Software Suite | A suite of tools (including CAMEO Chemicals and MARPLOT) used widely for planning for and responding to chemical emergencies, which can be relevant for assessing the real-world implications of chemical structures studied [16] [17]. |
Workflow Visualization: The Basin-Hopping Algorithm
The following diagram illustrates the iterative two-phase cycle of the basin-hopping algorithm.
Basin-Hopping Optimization Cycle
The Basin-Hopping (BH) algorithm is a powerful global optimization technique that has become indispensable in computational chemistry, physics, and drug development research. By transforming the complex energy landscape into a collection of basins, BH enables efficient navigation of potential energy surfaces (PES) to identify low-energy molecular configurations [12] [4]. This capability is particularly valuable in pharmaceutical research where predicting the stable conformations of drug molecules and their targets directly impacts therapeutic design. The algorithm's effectiveness stems from three core components working in concert: perturbation of atomic coordinates, local minimization to find basin bottoms, and the Metropolis criterion for accepting or rejecting new configurations [12] [2]. Understanding the precise implementation and troubleshooting of these components is essential for researchers applying BH to molecular structure prediction within their thesis investigations.
The BH algorithm follows a structured workflow that cycles through its three key components. The following diagram illustrates this iterative process:
Diagram Title: Basin-Hopping Algorithm Workflow
This workflow transforms the original potential energy surface into a staircase of local minima, effectively simplifying the search for the global minimum [4]. The algorithm's stochastic nature allows it to escape local minima while progressively focusing on lower-energy regions of the configuration space, making it particularly effective for complex molecular systems encountered in drug development research.
Essential Research Reagents and Computational Tools
The following table details key computational "reagents" and their functions in a typical BH investigation of molecular structures:
| Research Reagent | Function in Basin-Hopping Experiments |
|---|---|
| Local Optimizer (L-BFGS-B) | Minimizes energy from perturbed coordinates; finds local basin bottoms [9] [12] |
| Monte Carlo Step Generator | Creates random atomic displacements for PES exploration [9] [2] |
| Potential Energy Function | Calculates system energy (e.g., Lennard-Jones, molecular mechanics) [9] [2] |
| Metropolis Criterion | Stochastically accepts/rejects moves based on energy change and temperature [12] [4] |
| Adaptive Step Size Controller | Dynamically adjusts perturbation magnitude based on acceptance rates [9] |
| Parallel Processing Framework | Enables simultaneous evaluation of multiple trial structures [9] |
Troubleshooting Guide: Frequently Asked Questions
FAQ 1: Why does the algorithm fail to find the global minimum, getting stuck in high-energy regions?
Issue Analysis: This common problem in thesis research often stems from improper balance between exploration (perturbation) and exploitation (minimization).
Troubleshooting Steps:
- Adjust perturbation magnitude: The step size controls exploration capability. Implement an adaptive step size that monitors acceptance rates and adjusts to maintain a 30-50% acceptance rate [9].
- Modify temperature parameter: The Metropolis criterion temperature affects the probability of accepting uphill moves. Start with higher temperatures (allowing more exploration) and gradually reduce according to the schedule:
T_new = T_old * 0.99every 100 iterations [18]. - Increase iteration count: BH is stochastic and may require thousands of iterations for complex systems. For molecular clusters with 10-100 atoms, begin with at least 5,000-10,000 iterations [12] [18].
- Utilize parallel trials: Generate and minimize multiple (3-8) trial structures simultaneously at each step to improve sampling efficiency [9].
FAQ 2: Why are constraints and boundaries being violated during the optimization?
Issue Analysis: The perturbation step operates independently from the local minimizer, which may not respect your constraints if not properly configured [19] [20].
Troubleshooting Steps:
- Implement custom accept_test function: Create a bounds-checking function that rejects candidates violating constraints before Metropolis evaluation [20].
- Apply penalty functions: Modify the objective function to apply severe energy penalties when constraints are violated, naturally guiding the search away from invalid regions [20].
- Use constrained minimizers: For the local minimization phase, select algorithms that natively support constraints (e.g., COBYLA, SLSQP) through the
minimizer_kwargsparameter [19]. - Create custom step-taking routine: Develop a tailored
take_stepfunction that generates perturbations respecting your specific boundaries [19].
FAQ 3: Why is the algorithm converging too quickly to suboptimal solutions?
Issue Analysis: Premature convergence indicates insufficient exploration of the potential energy surface, often due to excessive "greediness" in the acceptance criteria.
Troubleshooting Steps:
- Increase temperature parameter: Higher temperatures in the Metropolis criterion increase the probability of accepting higher-energy configurations, preventing trapping in local minima. For energy scales of ~1-100 kJ/mol, start with kT values of 5-20 kJ/mol [4] [18].
- Implement mutation strategies: Occasionally apply larger perturbations or specific structural modifications (e.g., dihedral angle changes for molecular systems) to escape deep local minima [4].
- Utilize machine learning guidance: Incorporate similarity indices and clustering techniques to identify under-explored regions of the configuration space and direct perturbations accordingly [4].
- Restart from diverse points: Execute multiple independent BH runs from structurally distinct initial configurations and compare results [18].
FAQ 4: Why is the computation time prohibitively long for molecular systems?
Issue Analysis: Each BH iteration requires expensive energy and gradient calculations, creating bottlenecks particularly for ab initio potentials.
Troubleshooting Steps:
- Implement parallel candidate evaluation: Use multiprocessing frameworks to simultaneously evaluate 4-8 trial structures per iteration, achieving near-linear speedup on multi-core systems [9].
- Utilize efficient local minimizers: Select appropriate optimization algorithms (L-BFGS-B for large systems, BFGS for smaller ones) through the
minimizer_kwargsparameter [9] [12]. - Employ multi-level approaches: Use faster empirical potentials or machine learning force fields for initial exploration, then refine low-energy candidates with higher-level methods [9] [1].
- Adjust minimization tolerance: Loosen convergence criteria for early iterations (
gtol=0.1), tightening as the search progresses (gtol=0.001) [12].
Advanced Methodologies for Research Applications
Quantitative Parameter Optimization Table
The following table summarizes optimal parameter ranges for different research applications:
| Application Domain | Step Size Range | Temperature (kT) | Iterations | Parallel Trials |
|---|---|---|---|---|
| Lennard-Jones Clusters [9] | 0.1-0.5σ | 0.5-2.0ε | 2,000-10,000 | 4-8 |
| Molecular Conformers [4] | 0.5-1.5Å atomic displacement | 5-20 kJ/mol | 5,000-50,000 | 3-6 |
| Protein Structure Prediction [2] | 0.3-1.2Å Cα displacement | 50-200 K | 10,000-100,000 | 2-4 |
| Drug Molecule Docking [1] | 0.2-0.8Å + 15-45° rotation | 10-50 kJ/mol | 5,000-20,000 | 4-8 |
Machine Learning Enhancement Protocol
Advanced BH implementations can incorporate unsupervised machine learning techniques to improve PES exploration efficiency:
- Similarity Index Calculation: After each local minimization, compute the root-mean-square deviation (RMSD) or descriptor-based similarity between the new structure and all previously identified minima [4].
- Hierarchical Clustering: Periodically cluster all identified minima based on structural similarity to identify unexplored regions of the configuration space [4].
- Directed Perturbation: Bias future perturbations toward structurally under-sampled regions identified through clustering analysis [4].
- Transition State Identification: Use interpolation between identified minima to generate initial guesses for transition state searches, mapping complete reaction pathways [4].
This enhanced protocol significantly improves the comprehensiveness of PES mapping for thesis research requiring complete mechanistic understanding of molecular transformations.
The Basin-Hopping algorithm provides a robust framework for molecular structure prediction essential to pharmaceutical and materials research. By systematically addressing the common implementation challenges through the troubleshooting guidelines presented herein, researchers can optimize its performance for their specific thesis investigations. The continuous development of BH methodologies—including adaptive parameters, parallel implementation, and machine learning augmentation—ensures its ongoing relevance in computational chemistry and drug discovery.
Frequently Asked Questions (FAQs)
Q1: What is the Basin-Hopping algorithm and why is it used in global optimization? Basin-Hopping (BH) is a stochastic global optimization algorithm that transforms the potential energy surface into a collection of interpenetrating staircases, where each stair corresponds to a local minimum on the original landscape [4]. It combines random perturbation of coordinates with local minimization and Monte Carlo acceptance criteria to efficiently explore low-energy regions [2] [6]. BH is particularly valuable for overcoming large energetic barriers in multimodal optimization problems, making it well-suited for both atomic clusters and biomolecular structure prediction [2] [4].
Q2: How does the optimization of Lennard-Jones clusters relate to biomolecular structure prediction? Lennard-Jones (LJ) clusters serve as fundamental test cases for optimization algorithms due to their simple yet challenging energy landscapes with numerous local minima [21] [22]. The methodologies developed for LJ clusters, including Basin-Hopping, provided the foundation for tackling more complex biomolecular systems [2] [6]. Both problems share the challenge of navigating highly multimodal potential energy surfaces to find global minima, with LJ systems offering a computationally tractable proving ground for optimization techniques later applied to proteins [2].
Q3: What are the key limitations of the Basin-Hopping method? The principal limitations include: (1) Non-deterministic nature - identifying the global minimum is not guaranteed in finite searches [4]; (2) Potential kinetic trapping in local minima if simulation temperature is set too low [4]; (3) Performance dependence on the quality of the initial force field or potential energy function [4]; (4) Decreasing efficiency for large systems with simple Cartesian coordinate perturbations [2].
Q4: How have machine learning techniques augmented traditional Basin-Hopping approaches? Recent advancements integrate unsupervised machine learning concepts including similarity indices, hierarchical clustering, and multidimensional scaling to enhance BH searches [4]. These techniques help assess the uniqueness of local minima, guide potential energy surface exploration toward under-sampled regions, and facilitate the identification of transition states connecting minima [4]. This augmentation improves the efficiency of mapping complex molecular potential energy surfaces.
Troubleshooting Guides
Poor Convergence in Basin-Hopping Simulations
Symptoms:
- Algorithm fails to locate known global minima
- Excessive time spent sampling high-energy regions
- Repeated rediscovery of the same local minima
Possible Causes and Solutions:
| Cause | Diagnostic Steps | Solution |
|---|---|---|
| Inappropriate step size | Monitor acceptance rate; ideal rate is 0.3-0.5 [6] | Adjust perturbation magnitude; for benzene clusters, effective values are δR=1.5 Å, δΘ=0.75 rad [6] |
| Low simulation temperature | Check if algorithm is trapped in deep local minima | Increase temperature parameter to allow escape over higher barriers [4] |
| Insufficient sampling | Track unique minima encountered versus iterations | Extend simulation length; implement "significant structures" approach that updates geometry to local minimum [6] |
| Inefficient coordinate perturbation | For large systems, note decreased efficiency with Cartesian perturbations [2] | Implement collective moves or selective degree of freedom perturbation [4] |
Handling Biomolecular Complexity
Symptoms:
- Failure to identify biologically relevant conformations
- Computationally expensive energy evaluations
- Inability to locate transition states between minima
Solutions:
- Incorporate machine learning augmentation: Use similarity functions and clustering to identify unique conformations and guide sampling toward unexplored regions [4].
- Employ multi-level approaches: For large systems, leverage hierarchical structure by building larger systems from optimized smaller fragments [22].
- Implement enhanced sampling: Combine with techniques like umbrella sampling to confirm global minima are reached [2].
- Utilize specialized potentials: Apply bioinformatically-informed energy functions like the Associative Memory Hamiltonian to reduce landscape roughness [2].
Experimental Protocols
Standard Basin-Hopping Protocol for Structure Prediction
Detailed Procedure:
- Initialization: Generate random molecular geometry or start from known structure [4] [6].
- Iteration Cycle:
- Perturbation: Apply random changes to atomic coordinates (typically Cartesian) or selected degrees of freedom [2] [6].
- Minimization: Perform local energy minimization using gradient-based methods (e.g., truncated-Newton) to reach nearest local minimum [22] [6].
- Acceptance: Apply Monte Carlo criterion based on energy difference from previous minimum: accept if lower energy; for higher energy, accept with probability exp(-ΔE/kT) [4] [6].
- Termination: After fixed iterations or when no improvement occurs for specified number of steps [6].
Key Parameters:
- Step size (perturbation magnitude)
- Simulation temperature (for acceptance criterion)
- Maximum iterations
- Convergence threshold
Machine Learning-Augmented Basin-Hopping
Implementation Details:
- Similarity Assessment: Calculate distance metrics between minimized structures using root-mean-square deviation or other similarity functions [4].
- Clustering Analysis: Apply hierarchical clustering to identify families of related structures and detect sampling gaps [4].
- Guided Sampling: Bias subsequent perturbations toward under-explored regions of conformational space [4].
- Transition State Identification: Use interpolation between minima to generate guess geometries for transition state searches [4].
Research Reagent Solutions
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| Lennard-Jones Potential | Model interatomic interactions in noble gases and simple clusters [23] [21] | Standard test system for optimization algorithms; parameters (ε, σ) define energy and length scales [21] |
| Associative Memory Hamiltonian | Coarse-grained potential for protein structure prediction [2] | Incorporates bioinformatic data; uses memory proteins from database to guide folding [2] |
| Truncated-Newton Method | Local optimization algorithm [22] | Efficient for energy minimization steps; requires gradient information [22] |
| Similarity Functions | Quantify structural similarity between conformations [4] | Enables clustering and diversity maintenance; critical for ML-augmented BH [4] |
| Delaunay Triangulation | Identify atomic neighbors in clusters [22] | Used in multilevel methods to determine optimal atom placement [22] |
Performance Benchmarks and Data
Algorithm Performance Comparison
Table: Comparative Performance of Optimization Algorithms on Benchmark Problems [24]
| Algorithm | Convergence Speed | Success Rate (Global Minimum) | Scaling with System Size | Best Application Context |
|---|---|---|---|---|
| Basin-Hopping | Moderate | High for mild frustration | Moderate decrease for large systems | Mildly frustrated landscapes [2] |
| Basin-Hopping (Population) | Fast | High | Good | Multimodal problems [24] |
| Differential Evolution | Variable | Moderate | Good | General-purpose optimization [24] |
| CMA-ES | Fast | High | Good | Continuous domains [24] |
| Simulated Annealing | Slow | Moderate | Poor | Simple implementation needed [2] |
Historical Evolution of Methodologies
Table: Development of Basin-Hopping and Related Methods [2] [6] [4]
| Time Period | Methodological Focus | Key Advances | Representative Systems |
|---|---|---|---|
| 1990s | Basic BH algorithm | Transformation of PES; Monte Carlo with minimization [6] | Lennard-Jones clusters [6] |
| 2000s | Biological applications | Application to proteins; specialized potentials [2] | Small proteins; helical structures [2] |
| 2010s | Hybrid approaches | Multilevel methods; initial implementations for large systems [22] | Larger biomolecules [2] |
| 2020s | ML augmentation | Similarity analysis; hierarchical clustering [4] | Spectroscopy; ion mobility [4] |
The Critical Role of BH in Overcoming Limitations of Pure Local Search Methods
Basin-Hopping FAQ and Troubleshooting Guide
This guide provides targeted solutions for common challenges researchers face when implementing the Basin-Hopping (BH) algorithm for global optimization in computational chemistry and drug development.
Frequently Asked Questions
Q1: What is the core principle of the Basin-Hpping algorithm?
- A1: Basin-Hopping is a global optimization technique that transforms the potential energy surface by combining random Monte Carlo-like steps with local minimization. This process allows the search to escape local minima and explore different basins of attraction to find the global minimum [25]. It effectively maps the complex landscape into a collection of local minima, making the search for the global minimum more efficient [9].
Q2: Why is BH particularly effective for complex systems like Lennard-Jones clusters?
- A2: BH is highly effective for complex, high-dimensional landscapes with numerous local minima, such as Lennard-Jones clusters [9] [25]. Its two-phase method balances broad exploration of the configuration space with intensive exploitation of low-energy regions, allowing it to navigate the rugged energy landscapes that simpler local search methods cannot [9] [10].
Q3: How do I choose an appropriate step size for the perturbation step?
- A3: The step size is crucial and should be comparable to the typical separation between local minima in your system [10]. An optimal strategy is to use an adaptive step size that is dynamically adjusted to maintain a target acceptance rate (e.g., 50%). This automatically balances exploration and refinement during the search [9] [10].
Q4: What is the role of the "temperature" (T) parameter in the acceptance criterion?
- A4: The temperature parameter in the Metropolis acceptance criterion controls the probability of accepting uphill moves. If
func(xnew) < func(xold), the step is always accepted. Otherwise, it is accepted with probabilityexp( -(func(xnew) - func(xold)) / T )[10]. For best results,Tshould be comparable to the typical function value difference between local minima [10]. SettingT=0turns the algorithm into Monotonic Basin-Hopping, where all uphill moves are rejected [10].
- A4: The temperature parameter in the Metropolis acceptance criterion controls the probability of accepting uphill moves. If
Q5: How can I accelerate BH calculations for computationally expensive systems?
- A5: A highly effective strategy is parallel evaluation of candidate structures. Multiple perturbed configurations can be minimized concurrently on different CPU cores, significantly reducing wall-clock time. Research shows nearly linear speedup can be achieved for up to eight concurrent local minimizations [9].
Troubleshooting Common Experimental Issues
| Problem Symptom | Potential Cause | Diagnostic Steps | Solution |
|---|---|---|---|
| Search stagnates in a local minimum. | Step size is too small; temperature is too low. | Check acceptance rate history. A very low rate suggests poor exploration. | Increase the step size or the T parameter adaptively [9] [10]. |
| Search behaves randomly without convergence. | Step size is too large; temperature is too high. | Check energy trace for large, non-improving fluctuations. | Decrease the step size or the T parameter [10]. |
| Computation time is prohibitively long. | Sequential processing; expensive local minimizations. | Profile code to identify bottlenecks (likely the local minimization step). | Implement parallel evaluation of multiple trial structures [9]. |
| Algorithm fails to find known global minima on benchmark systems (e.g., LJ38). | Inadequate sampling; weak local minimizer. | Run from multiple random starting points (x0) to check consistency. |
Increase the number of iterations (niter); use a more robust local minimization method (e.g., L-BFGS-B) [9] [10]. |
Key Experimental Parameters and Default Values
The following parameters are critical for controlling the behavior and performance of the BH algorithm. The table below summarizes typical values based on the SciPy implementation and recent research [9] [10].
| Parameter | Description | Typical/Default Value | Impact on Search |
|---|---|---|---|
niter |
Number of basin-hopping iterations. | 100 [10] | More iterations increase the probability of success. |
T |
"Temperature" for the Metropolis criterion. | 1.0 [10] | Higher T accepts more uphill moves, aiding escape from local minima. |
stepsize |
Maximum step size for random displacement. | 0.5 [10] | Larger steps promote exploration; smaller steps favor local refinement. |
target_accept_rate| Target for the adaptive step size mechanism. |
0.5 [10] | Dynamically adjusts stepsize to maintain this acceptance rate. |
|
interval |
Number of iterations between step size updates. | 50 [10] | Controls how frequently the adaptive strategy is applied. |
niter_success |
Stop if the best minimum doesn't change for this many iterations. | None | Can be set to trigger automatic early stopping. |
Detailed Experimental Protocol: Adaptive Basin-Hopping
This protocol outlines the steps for running an adaptive Basin-Hopping search for a molecular cluster, using a Lennard-Jones potential as an example [9].
Objective: Locate the global minimum energy structure of an (N)-atom Lennard-Jones cluster. Software Tools: Python, SciPy library, NumPy.
Initial Structure Preparation:
- Generate or obtain an initial guess for the cluster geometry. This can be a random configuration or a known structure.
- Save the initial coordinates in a format readable by the script (e.g., a
.moldenfile).
Algorithm Configuration:
- Local Minimizer: Configure the local minimization step. The L-BFGS-B algorithm is often a robust choice [9].
- BH Parameters: Set the core parameters:
- Number of iterations (
niter): Start with 100-500 for testing. - Temperature (
T): Set to 1.0 as a starting point. - Initial step size (
stepsize): Set to 0.5. - Adaptive parameters: Set
target_accept_rate=0.5andinterval=50[10].
- Number of iterations (
Execution:
- Execute the BH algorithm. The process for each iteration is: a. Perturbation: Apply a random displacement to the current coordinates. b. Local Minimization: Use the local minimizer to find the nearest local minimum for the perturbed structure. c. Acceptance: Apply the Metropolis criterion to decide whether to accept the new minimum. d. Adaptation: Periodically, adjust the step size based on the recent acceptance rate to maintain the target [9] [10].
Analysis:
- Plot the energy trace of accepted and trial minima to visualize the search progression.
- Identify the lowest-energy structure found and analyze its geometry.
- Compare the result against known global minima for validation (e.g., from the Cambridge Cluster Database).
The Scientist's Toolkit: Essential Research Reagents
| Item | Function in the Experiment |
|---|---|
| Local Minimizer (e.g., L-BFGS-B) | A deterministic algorithm that finds the nearest local minimum from a given starting configuration. This is the core "refinement" step in BH [9]. |
| Potential Energy Function (e.g., Lennard-Jones) | A mathematical function that calculates the total energy of a system given the coordinates of its atoms. This is the "cost function" to be minimized [9]. |
| Monte Carlo Step Generator | A routine that produces random perturbations (e.g., atomic displacements) to create new trial structures, enabling the global exploration of the energy landscape [10]. |
| Adaptive Step Size Controller | A feedback mechanism that dynamically adjusts the magnitude of random perturbations based on the recent acceptance rate, optimizing the balance between exploration and refinement [9] [10]. |
| Parallel Processing Framework | A computational environment (e.g., Python's multiprocessing.Pool) that allows multiple trial structures to be minimized simultaneously, drastically reducing the time required for convergence [9]. |
Workflow and Algorithm Diagrams
Diagram Title: Basin-Hopping Algorithm Core Workflow
Diagram Title: Adaptive Step Size Control Logic
Implementing Basin-Hopping: From Theory to Practice in Drug Discovery
Step-by-Step Workflow of a Standard Basin-Hopping Algorithm
A Researcher's FAQ on Basin-Hopping
What is the core principle behind the basin-hopping algorithm? Basin-hopping is a global optimization technique that transforms the energy landscape of a problem into a collection of basins of attraction. It explores this transformed landscape by iteratively performing random perturbations (hopping) to escape local minima, followed by local minimization to find the bottom of each new basin [12] [25].
How does the algorithm decide whether to accept a new local minimum? The acceptance of a new solution, even if it is worse than the current one, is typically governed by a stochastic criterion. The most common is the Metropolis criterion, where the probability of accepting a new solution with energy ( E{\text{new}} ) and a current solution with energy ( E{\text{old}} ) is ( \exp(-(E{\text{new}} - E{\text{old}}) / T) ), where ( T ) is a temperature-like parameter controlling the acceptance of uphill moves [26] [27].
What are the most critical hyperparameters to tune for a successful experiment?
The key hyperparameters are the number of iterations (niter), the step size for perturbations (stepsize), and the temperature (T) in the acceptance criterion [12] [27]. The step size should be chosen to be a reasonable fraction of your problem's characteristic length scales to effectively jump between basins [12].
Which local minimizer should I use? The choice can depend on your specific problem. The L-BFGS-B algorithm is a common and efficient default for smooth, bounded problems [12]. For problems where derivative information is unavailable or unreliable, the Nelder-Mead simplex method can be a good alternative [12].
The Scientist's Toolkit: Essential Components for a Basin-Hopping Experiment
| Component | Function & Description |
|---|---|
| Objective Function | The core of the optimization; a function that takes coordinates as input and returns a scalar value (e.g., energy) to be minimized [28]. |
| Local Minimizer | An algorithm (e.g., L-BFGS-B, BFGS, Nelder-Mead) responsible for finding the local minimum from a given starting point [12] [26]. |
| Perturbation Method | A strategy (e.g., random displacement) for modifying the current best coordinates to generate a new starting point for the next local search [12] [26]. |
| Acceptance Test | A criterion (e.g., Metropolis test) to decide whether to accept or reject a new local minimum based on its energy and the current system "temperature" [26] [27]. |
Workflow of the Standard Basin-Hopping Algorithm
The following diagram illustrates the iterative cycle of perturbation, local minimization, and acceptance that defines the basin-hopping method.
Detailed Experimental Protocol and Parameter Configuration
To implement a basin-hopping experiment, follow this structured protocol, paying close attention to the configuration of key parameters.
1. Initialization
- Define the Objective Function: Code your function ( f(\vec{x}) ) to be minimized, where ( \vec{x} ) is the vector of coordinates [28].
- Select an Initial Guess: Choose a starting point ( \vec{x}_0 ). This can be a random point within the domain of interest or an informed guess based on prior knowledge [12].
- Configure Hyperparameters: Set the algorithm's control parameters. The table below provides a guideline.
| Parameter | Description | Recommended Starting Value |
|---|---|---|
Number of Iterations (niter) |
Total basin-hopping steps; determines runtime [12]. | 100 - 1000 (problem-dependent) |
Step Size (stepsize) |
Maximum perturbation magnitude; critical for jumping basins [12] [27]. | ~5% of the problem's characteristic domain size [12] |
Temperature (T) |
Controls acceptance probability of worse solutions; higher T encourages exploration [26] [27]. | 1.0 (often requires tuning) |
| Local Minimizer | Algorithm for local optimization (e.g., 'L-BFGS-B', 'BFGS', 'Nelder-Mead') [12]. |
'L-BFGS-B' |
2. Iteration Cycle
For each iteration from 1 to niter:
- Perturbation: Generate a trial coordinate ( \vec{x}{\text{trial}} = \vec{x}{\text{current}} + \Delta \vec{x} ), where ( \Delta \vec{x} ) is a random displacement. The
stepsizeparameter typically bounds this displacement [12] [27]. - Local Minimization: From ( \vec{x}{\text{trial}} ), perform a local optimization to find a new local minimum ( \vec{x}{\text{new}} ) with value ( E{\text{new}} = f(\vec{x}{\text{new}}) ) [12] [26].
- Acceptance/Rejection: Apply the acceptance test (e.g., Metropolis criterion) to decide whether to accept ( \vec{x}{\text{new}} ). If ( E{\text{new}} < E{\text{current}} ), always accept. Otherwise, accept with probability ( \exp(-(E{\text{new}} - E_{\text{current}}) / T) ) [26] [27].
- Update: If the new minimum is accepted, set ( \vec{x}{\text{current}} = \vec{x}{\text{new}} ) and ( E{\text{current}} = E{\text{new}} ).
3. Termination
The algorithm terminates after completing the specified niter cycles. The best-found solution ( \vec{x}{\text{current}} ) and its value ( E{\text{current}} ) are returned as the putative global minimum [12].
Troubleshooting Common Experimental Issues
Problem: The algorithm converges to a local minimum, not the global minimum.
- Cause & Solution: The temperature
Tmay be too low, preventing escapes from deep local minima. The step size may be too small to jump to a new basin. IncreaseTand/orstepsizeto promote more exploration. Also, ensureniteris large enough for the landscape to be sufficiently sampled [12] [27].
Problem: The optimization is too slow.
- Cause & Solution: The local minimizer may be inefficient. For large systems, ensure you are using a fast, gradient-based minimizer like L-BFGS-B instead of derivative-free methods like Nelder-Mead. The objective function or its gradient may be computationally expensive; profile your code to identify bottlenecks [12] [28].
Problem: The algorithm is unstable and fails to converge.
- Cause & Solution: The step size might be too large, causing the algorithm to behave like a random search. Implement an adaptive step size that adjusts based on the observed acceptance ratio to meet a target (e.g., 0.5) [27].
Common Error 1: Algorithm Does Not Find the Global Minimum
Problem: The algorithm returns a local minimum instead of the global minimum.
Root Cause: Basin-hopping is a stochastic algorithm, and finding the global minimum is not guaranteed. It can get trapped in a local basin, especially if niter is too low, the stepsize is inappropriate, or the "temperature" T is poorly calibrated [11] [18].
Solution:
- Increase Iterations: Significantly increase the
niterparameter (e.g., from 100 to 1000 or more) to allow for more exploration [11]. - Adjust Step Size: The
stepsizeshould be comparable to the typical distance between local minima in your parameter space. If it's too small, the algorithm cannot jump to new basins; if too large, it becomes inefficient [29]. - Calibrate Temperature (
T): The temperatureTin the Metropolis acceptance criterion should be comparable to the typical function value difference between local minima. A higher T allows the algorithm to accept uphill moves more easily, facilitating escape from local minima [29]. - Use a Random Seed: For reproducibility and to test robustness, run the algorithm multiple times with different random seeds (
rngparameter) [30].
Common Error 2: Algorithm Ignores User-Defined Bounds
Problem: The solution provided by basinhopping violates the desired bounds for the parameters.
Root Cause: The bounds are only enforced in the high-level acceptance test. The local minimizer (e.g., "BFGS") called at each step is often unconstrained and knows nothing about these bounds. It can follow the gradient straight out of the allowed region [20].
Solution:
- Use a Bounded Local Minimizer: In the
minimizer_kwargsparameter, specify a local minimization method that inherently respects bounds, such as"L-BFGS-B", and provide theboundsto it [30]. - Penalize the Objective Function: Modify your objective function to return a very large (penalized) value for inputs outside the bounds, which naturally steers the minimizer away from invalid regions [20].
Common Error 3: Inconsistent Results with Different Settings
Problem: The algorithm returns different results when the local minimizer method or initial guess (x0) is changed.
Root Cause: This is expected behavior. The performance of the basin-hopping algorithm is highly sensitive to the choice of the local minimizer, the initial point, and its inherent random component [30]. Different local minimizers may converge to different local minima from the same starting point after a perturbation.
Solution:
- Benchmark Minimizers: Test different local minimization methods (e.g.,
"BFGS","Nelder-Mead","L-BFGS-B") via theminimizer_kwargsto find the most effective one for your specific function [12] [30]. - Multiple Runs: Run the algorithm multiple times from different starting points to build confidence that you have found the global minimum [29] [18].
Frequently Asked Questions (FAQs)
FAQ 1: What is the meaning of the output parameters likenfev,nit, andminimization_failures?
The OptimizeResult object returned by basinhopping contains several key metrics [31] [32]:
| Parameter | Description |
|---|---|
nfev |
Total number of function evaluations. |
nit |
Number of basin-hopping iterations completed. |
njev |
Number of Jacobian (gradient) evaluations. |
minimization_failures |
Number of times the local minimizer failed to converge. |
fun |
Value of the objective function at the solution. |
x |
Solution array (the optimized parameters). |
Note that nfev is typically much larger than nit because each iteration (nit) involves many function evaluations during the local minimization step [31].
FAQ 2: How can I increase my confidence that the found minimum is truly global?
No stochastic algorithm can guarantee it has found the global minimum. However, you can increase your confidence by [18]:
- Running the algorithm for a long time (high
niter). - Using a large value for
niter_successto stop only if the best candidate remains unchanged for many iterations. - Running the algorithm multiple times from diverse starting points (
x0) and with different random seeds (rng). - For some problems, a brute-force approach (
scipy.optimize.brute) can be used on a coarse grid to identify promising regions forbasinhoppingto explore.
FAQ 3: What is the role of the "temperature" parameterT?
The temperature T controls the acceptance of uphill moves via the Metropolis criterion. A step is always accepted if it finds a lower function value. If the function value increases by ΔE, the step is accepted with a probability of exp(-ΔE / T) [29].
- A high T makes the algorithm more likely to accept worse solutions, helping it escape local minima.
- A low T makes the algorithm more greedy, only accepting moves that improve the solution.
- If
T=0, the algorithm becomes Monotonic Basin-Hopping, where any uphill move is rejected [29]. For best results,Tshould be comparable to the typical difference in function values between local minima.
Experimental Protocol: Optimizing a Multimodal Function
This protocol outlines how to use the basin-hopping algorithm to find the global minimum of the Ackley function, a common benchmark problem with many local minima.
1. Define the Objective Function The Ackley function is defined in Python as follows [12]:
2. Configure and Execute the Basin-Hopping Algorithm
3. Expected Workflow The following diagram illustrates the basin-hopping algorithm's iterative cycle:
4. Key Research Reagent Solutions
| Item | Function in Optimization | Typical Configuration / Notes |
|---|---|---|
| Local Minimizer | Finds the lowest point in the current basin of attraction. | "L-BFGS-B" (with bounds), "BFGS", "Nelder-Mead" [12] [30]. |
| Step Size | Controls the maximum random perturbation to jump to new basins. | Should be ~5% of the domain size (e.g., 0.5 for a [-5,5] domain) [12]. |
Temperature (T) |
Controls the probability of accepting worse solutions to escape local minima. | Should be set to the typical energy (function value) difference between local minima [29]. |
Random Number Generator (rng) |
Ensures reproducible results for stochastic steps. | Use a fixed seed for debugging and reproducibility [29] [30]. |
Frequently Asked Questions
Q1: What is the most common reason for the algorithm getting stuck in a poor local minimum?
- A: This is often due to a step size that is too small, preventing the search from escaping the current basin of attraction. Alternatively, a temperature parameter that is too low can cause the algorithm to reject any moves that temporarily increase the objective function value, even if they might lead to a better basin.
Q2: How should I interpret the "temperature" parameter (T)? It doesn't seem to relate to physical temperature.
- A: The temperature
Tin the Metropolis acceptance criterion is an algorithmic parameter. For best results,Tshould be comparable to the typical separation in function value between the local minima in your problem landscape. It has no direct relation to a physical temperature unless your objective function is a physical energy [33] [34].
- A: The temperature
Q3: Does the step size remain fixed throughout the optimization?
- A: No, by default, the step size is often adjusted adaptively to meet a target acceptance ratio. The algorithm will periodically increase or decrease the step size (e.g., by multiplying or dividing by a factor like 0.9) to try and maintain an optimal balance between exploration and exploitation [35] [34].
Q4: My optimization is taking too long. How can I speed it up?
Q5: How do I know if I have run enough iterations?
- A: There is no universal answer, as it depends on the complexity of your problem's energy landscape. A good practice is to run the algorithm multiple times from different starting points and see if it consistently finds the same solution. Some implementations also allow you to set a stop condition (
niter_success) if the global minimum candidate remains unchanged for a specified number of iterations [34].
- A: There is no universal answer, as it depends on the complexity of your problem's energy landscape. A good practice is to run the algorithm multiple times from different starting points and see if it consistently finds the same solution. Some implementations also allow you to set a stop condition (
Troubleshooting Guides
Problem: Poor Performance on Rugged, High-Dimensional Landscapes
- Description: The algorithm fails to locate good minima, particularly in complex, high-dimensional search spaces common in fields like drug development (e.g., molecular structure prediction).
- Solution: Augment the standard Basin-Hopping algorithm with techniques from unsupervised machine learning.
- Experimental Protocol:
- Define a Similarity Function: During the BH search, compute a similarity metric
d(A, B)between all saved local minima. This function should be non-negative, symmetric, and yield zero only for identical structures [4]. - Apply Hierarchical Clustering: Use the matrix of similarities to perform hierarchical clustering on the found minima. This creates a dendrogram that maps the explored regions of the potential energy surface (PES) [4].
- Analyze and Guide Search: Identify large clusters that have been thoroughly explored and regions that are sparsely sampled. Use this information to bias future random perturbations towards unexplored regions, ensuring a more comprehensive search of the landscape [4].
- Define a Similarity Function: During the BH search, compute a similarity metric
Problem: Inefficient Search and Slow Convergence
- Description: The optimization process requires an excessively long time to find a satisfactory solution.
- Solution: Systematically configure the core hyperparameters based on your problem's characteristics.
- Experimental Protocol:
- Characterize Your Objective Function: Perform a preliminary analysis to understand the approximate scale and typical separations between local minima in your function.
- Configure Hyperparameters: Refer to the following table for specific guidance. The values are drawn from common practices in chemical physics and general global optimization [36] [27] [34].
| Hyperparameter | Description & Function | Recommended Value / Strategy | Rationale |
|---|---|---|---|
| Step Size | Maximum displacement for random perturbation of coordinates. Defines "jump" magnitude to new basins [36]. | • Start with ~2.5-5% of the parameter domain size (e.g., 0.5 for [-5,5] domain) [12].• Use adaptive step size (often the default) [35]. | A small step size causes trapping; too large prevents local refinement. Adaptive control maintains efficient exploration. |
Iterations (niter) |
Total number of basin-hopping steps (perturbation + local minimization) [36]. | • Start with 100-200 for simple problems [36].• Use 10,000+ for complex systems like molecular clusters [27]. | More iterations allow exploration of more basins. Complex systems require extensive sampling for reliable results. |
Temperature (T) |
Controls acceptance of worse solutions via Metropolis criterion: exp( -(f_new - f_old) / T ) [33] [34]. |
Set comparable to the typical function value difference between local minima [33]. | A high T accepts most moves (like random search). A low T only accepts improving moves (greedy). A well-chosen T helps tunnel through barriers. |
The logical workflow for applying these concepts is summarized in the following diagram:
Problem: Algorithm Becomes Trapped in a Single Basin
- Description: The search appears to converge quickly, but the solution is clearly a suboptimal local minimum.
- Solution: Increase the step size and/or temperature to encourage exploration.
- Experimental Protocol:
- Increase Step Size: Double the initial step size. If you are using an adaptive method, check the final adapted step size from a previous run; if it is very small, it indicates the initial value was too large and was constantly reduced. In this case, try a larger initial value to promote bigger jumps [35].
- Increase Temperature: Multiply the temperature
Tby a factor of 10. This makes the algorithm more likely to accept transitions that temporarily increase the function value, allowing it to escape the current basin [33] [34]. - Use "Jump" Moves: Some advanced implementations offer a "jump" feature after a certain number of consecutive rejections, providing a more global search to escape the current basin [27].
The Scientist's Toolkit: Research Reagent Solutions
The following table details key computational "reagents" essential for a successful Basin-hopping experiment in scientific research.
| Item / Solution | Function / Role in the Experiment |
|---|---|
| Local Minimizer (e.g., L-BFGS-B, Nelder-Mead) | The algorithm used to descend to the bottom of a local basin after each perturbation. The choice can affect both speed and robustness of the overall search [36] [12]. |
| Similarity Metric & Clustering | A function to quantify the difference between two solution structures (e.g., RMSD for molecules). Used to map the explored landscape and guide the search away from over-explored regions [4]. |
| Metropolis Criterion | The probabilistic rule (based on temperature T) that determines whether a new, higher-energy solution is accepted, allowing the search to escape local minima [33] [34]. |
| Adaptive Step Size Controller | A mechanism that automatically adjusts the perturbation step size during the run to maintain a target acceptance rate, improving optimization efficiency [35] [34]. |
Selecting an Effective Local Minimizer (L-BFGS-B, Nelder-Mead)
Frequently Asked Questions
1. What is the fundamental difference between the L-BFGS-B and Nelder-Mead algorithms?
The core difference lies in their use of derivative information. L-BFGS-B is a second-order method that uses gradient information (the first derivative) to guide its search and can efficiently handle bound constraints on variables [37] [38]. In contrast, Nelder-Mead is a pattern search algorithm that does not require or use any derivative information, making it suitable for problems where the gradient is unknown, noisy, or non-differentiable [39].
2. My optimization is stuck. How can I troubleshoot a poorly performing minimizer?
First, verify the differentiability of your objective function. Using round(), int(), or abs() in your function can create discontinuities that break gradient-based methods like L-BFGS-B [40]. For such functions, switch to a derivative-free method like Nelder-Mead. Second, check if your problem is multimodal (has many local minima). Both L-BFGS-B and Nelder-Mead can get stuck in local minima [39] [41]. In the context of Basin Hopping, this is mitigated by the algorithm's stochastic perturbations and Metropolis acceptance criterion, which help escape shallow minima [9].
3. The L-BFGS-B solver converges to a poor solution or fails. What could be wrong?
This can occur due to recent updates in scientific libraries. A known issue was observed between SciPy versions 1.14 and 1.15, where the L-BFGS-B implementation showed worse behavior on some test functions, converging to a non-optimal point with fewer function evaluations [42]. If you encounter this, consider:
- Checking your SciPy version.
- Providing an explicit gradient function via the
jacargument to ensure accuracy [37] [38]. - Trying a different starting point.
4. When should I prefer Nelder-Mead in my Basin Hopping research?
Nelder-Mead is advantageous in several scenarios relevant to computational chemistry and drug development:
- Noisy or experimental data: When your objective function (e.g., an energy potential) is inherently noisy [39].
- Non-differentiable functions: When the potential energy surface is complex or not smoothly differentiable.
- Rugged landscapes: While it can get stuck, its lack of reliance on gradients can sometimes be beneficial on pathological surfaces, though global strategies like Basin Hopping are still recommended [39] [9].
Optimizer Characteristics and Selection Guide
The following table summarizes the key attributes of the L-BFGS-B and Nelder-Mead algorithms to aid in selection.
| Feature | L-BFGS-B | Nelder-Mead |
|---|---|---|
| Algorithm Type | Quasi-Newton (Second-Order) [37] [38] | Pattern Search (Zero-Order) [39] |
| Derivative Use | Requires gradients (Uses first derivative) [37] [38] | Derivative-free [39] |
| Constraint Handling | Supports bound constraints [37] | Unconstrained |
| Typical Application | Smooth, convex functions [37] [38] | Non-differentiable or noisy objective functions [39] |
| Memory & Performance | Efficient for large problems (limited-memory) [37] | Simpler, but can require more function evaluations [39] |
| Common Pitfalls | Fails on non-differentiable functions [40]; Sensitive to implementation changes [42] | Can get stuck in local optima; May show erratic convergence [39] [41] |
Experimental Protocol for Local Minimization
The workflow below integrates local minimizers into a broader global optimization strategy, such as the Basin Hopping method used for finding low-energy cluster configurations in drug development and materials science [9].
Detailed Methodology
Initialization: Begin with an initial molecular or atomic cluster structure, typically read from a file format like Molden that contains Cartesian coordinates [9].
Perturbation: Apply a random displacement to the current structure's atomic coordinates to escape the current local minimum. The step size for this perturbation can be adaptive, dynamically adjusted to maintain a 50% acceptance rate for efficient exploration [9].
Local Minimization: This is the critical step where a local minimizer like L-BFGS-B or Nelder-Mead is employed.
Objective Function: Evaluate the potential energy of the perturbed structure using a defined potential (e.g., Lennard-Jones for clusters or a molecular mechanics force field for drug-like molecules). The function to minimize is the total energy, ( E ) [9]. For a Lennard-Jones cluster, this is given by:
( E{LJ} = 4\epsilon \sum{i
where ( r_{ij} ) is the distance between atoms ( i ) and ( j ), and ( \epsilon ) and ( \sigma ) are potential parameters [9].
- Minimizer Execution: Pass the objective function and the flattened coordinate array to the minimizer. For L-BFGS-B, you can also provide the gradient of the potential for faster, more reliable convergence [37].
Metropolis Criterion: Decide whether to accept the newly minimized structure. The acceptance probability is ( p = \exp(-\Delta E / T) ), where ( \Delta E ) is the energy difference between the new and old structures, and ( T ) is an effective temperature (often set to 1.0). This stochastic rule allows the search to escape higher-energy local minima [9].
Convergence Check: Repeat steps 2-4 until a convergence criterion is met (e.g., a maximum number of steps is reached or no lower energy is found for a significant number of iterations).
Performance Notes
- Parallel Evaluation: To reduce computational time, multiple perturbed trial structures can be minimized concurrently (in parallel) at each Basin Hopping step. The lowest-energy result is then selected for the Metropolis step [9].
- Algorithm Choice: L-BFGS-B is often preferred for its speed on smooth landscapes, while Nelder-Mead is a robust fallback for noisy or non-differentiable potentials [39] [37].
Research Reagent Solutions
The following table lists key computational components used in optimization experiments for structural prediction.
| Component | Function | Implementation Example |
|---|---|---|
| L-BFGS-B Minimizer | Efficiently finds local minima for smooth functions with bound constraints [37]. | scipy.optimize.minimize(obj_func, x0, method='L-BFGS-B', jac=grad_func, bounds=bnds) |
| Nelder-Mead Minimizer | Finds local minima without requiring derivatives, useful for noisy or complex landscapes [39]. | scipy.optimize.minimize(obj_func, x0, method='nelder-mead') |
| Lennard-Jones Potential | A classic model for pairwise atomic interactions in noble gas clusters and materials [9]. | 4 * epsilon * ((sigma/r)12 - (sigma/r)6) |
| Rosenbrock Function | A standard non-convex test function for benchmarking optimization algorithms [37]. | f(x) = (1 - x[0])2 + 100 * (x[1] - x[0]2)2 |
| Basin Hopping Wrapper | Global optimization routine that combines random perturbation with local minimization to find low-energy states [9]. | Custom scripts or libraries like scipy.optimize.basinhopping |
Troubleshooting Guide: Common Basin-Hopping Issues
This guide addresses frequent challenges encountered when using the Basin-Hopping (BH) algorithm for identifying low-energy conformers of drug-like molecules.
| Problem Description | Possible Causes | Recommended Solutions |
|---|---|---|
| Slow Convergence | Step size too large/small, inadequate local minimizer, insufficient iterations [9] [28]. | Implement adaptive step size control to maintain ~50% acceptance rate [9]. Use efficient, gradient-based minimizers (e.g., L-BFGS-B) [9] [43]. |
| Trapping in Local Minima | Insufficient thermal energy (kBT), lack of search diversity [9] [44]. | Utilize parallel trials: generate/multiple perturbed structures and select the best after minimization [9]. Apply stronger perturbation moves or "kick" procedures to escape deep minima [44]. |
| High Computational Cost | Expensive energy evaluations (e.g., DFT), inefficient force calculations, serial execution [9] [28]. | Parallelize local minimization steps across multiple CPU cores [9]. Replace ab initio calculations with machine-learned potentials for initial screening [9]. Vectorize force/energy calculations [28]. |
| Poor Quality Final Structures | Inadequate sampling of conformational space, premature termination [9]. | Combine BH with other algorithms (e.g., Molecular Dynamics) for broader sampling [44]. Run multiple independent BH runs from different random seeds. Increase number of BH steps. |
| Gradient Calculation Errors | Numerical instability, discontinuities in the potential energy surface (PES). | Use analytical gradients where possible [28]. Switch to a minimizer robust to numerical noise. Check for invalid molecular geometries after perturbation. |
Frequently Asked Questions (FAQs)
Q1: What is the key advantage of the Basin-Hopping method for conformer search compared to a systematic search? Basin-Hopping is highly efficient for navigating the complex, high-dimensional Potential Energy Surfaces (PES) of drug-like molecules. Instead of exhaustively sampling all angles, it uses Monte Carlo moves combined with local minimization to "hop" between energy basins, focusing computational resources on low-energy regions and effectively bypassing high-energy transition states [9] [44]. This makes it feasible to study flexible molecules with many rotatable bonds.
Q2: How do I choose an appropriate step size for the random atomic displacements? The optimal step size balances exploration and exploitation. A very small step size leads to trapping in a single basin, while a very large one reduces acceptance rates. The recommended strategy is to use an adaptive step size: monitor the acceptance rate over a window of steps (e.g., 50) and adjust the step size to achieve a target acceptance rate of approximately 50% [9].
Q3: Can Basin-Hopping be used with high-level quantum mechanics (QM) methods like Density Functional Theory (DFT)? Yes, but the computational cost can be prohibitive for large-scale searches. A practical approach is to use a multi-level strategy. Perform the initial BH sampling with a fast, lower-level method (like a forcefield or semi-empirical QM) to identify promising low-energy candidate structures. These candidates can then be refined using more accurate (and expensive) methods like DFT [9].
Q4: My system gets stuck in a specific local minimum. How can I encourage the search to escape? Implementing a parallel trial strategy can significantly reduce trapping. Instead of generating one new candidate per BH step, generate multiple (e.g., 4-8) independent perturbed structures, minimize them in parallel, and use the lowest energy result for the Metropolis acceptance test. This increases the chance of finding a lower path out of the current minimum [9].
Q5: How does the Metropolis criterion help in finding the global minimum?
The Metropolis criterion is crucial for global optimization. By sometimes accepting moves that increase the energy (with a probability of exp(-ΔE/kBT)), the algorithm can escape from local minima and explore other regions of the PES. The effective temperature parameter T acts as a control: higher T encourages more exploration, while lower T favors refinement of low-energy structures [9] [28].
Experimental Protocol: Basin-Hopping for Drug-like Molecules
The following provides a detailed methodology for a BH-based conformer search, as referenced in current literature [9] [44] [28].
The diagram below illustrates the iterative BH cycle with parallel trials, a key acceleration technique [9].
Step-by-Step Procedure
System Initialization:
- Generate an initial 3D structure for the drug-like molecule. This can be a rough 3D model from a 2D drawing or an extended conformation.
- Set BH parameters: initial step size (e.g., 0.1-0.3 Å), temperature
Tfor Metropolis criterion (e.g., 1.0-3.0 in reduced units), number of parallel trials per step (e.g., 4-8), and total number of BH iterations [9].
Iterative BH Cycle:
- Perturbation: Apply a random displacement to the atomic coordinates of the current best structure. The displacement is typically within a cube of side
2 * step_size[9]. - Parallel Local Minimization: For each of the
nparallel trials, take the perturbed structure and perform a local energy minimization. Use a reliable algorithm like L-BFGS-B which is efficient for large systems [9] [43]. This step maps the structure to the bottom of its current energy basin. - Selection: From the
nminimized structures, select the one with the lowest potential energy. - Metropolis Acceptance: Compare the energy of this new candidate (
E_new) with the current best structure (E_current). Accept the new candidate if: - Step Size Adaptation: Every 10-50 steps, calculate the recent acceptance rate. If it is below a target (e.g., 0.5), decrease the step size; if above, increase it [9].
- Perturbation: Apply a random displacement to the atomic coordinates of the current best structure. The displacement is typically within a cube of side
Termination and Analysis:
- Stop the search after a predefined number of steps, when no new low-energy minima are found for a long period, or when a specific energy threshold is reached.
- Cluster the accepted structures from the BH run to identify unique, low-energy conformers.
- The structure with the absolute lowest energy is the putative global minimum conformer.
The Scientist's Toolkit: Essential Research Reagents & Solutions
The following table details key computational "reagents" and tools essential for conducting a Basin-Hopping study.
| Item/Software | Function/Benefit | Application Note |
|---|---|---|
| L-BFGS-B Optimizer | A quasi-Newton local minimizer ideal for large systems; uses estimated Hessian for fast convergence [9] [43]. | Preferred for its memory efficiency and robustness. Available in scipy.optimize. Critical for the local minimization step. |
| Lennard-Jones / Forcefields | Fast, empirical potentials for initial sampling and testing. The LJ potential is a standard benchmark [9]. | Use for proof-of-concept and method validation. Switch to more specific forcefields (GAFF, CGenFF) for organic/drug-like molecules. |
| Multiprocessing Pool (Python) | Enparallel evaluation of trial structures, drastically reducing wall-clock time [9]. | Implement using multiprocessing.Pool. Near-linear speedups are achievable for 4-8 concurrent processes [9]. |
| Metropolis Criterion | The core logic allowing the search to escape local minima by sometimes accepting uphill moves [9] [28]. | The effective temperature T is a critical parameter. Higher T increases exploration. |
| Adaptive Step Size | Dynamically adjusts perturbation magnitude to maintain a healthy ~50% acceptance rate, improving efficiency [9]. | Prevents the search from becoming trapped (step too small) or behaving randomly (step too large). |
| Machine-Learned Potentials (MLPs) | Neural network potentials can provide near-quantum accuracy at a fraction of the computational cost of DFT [9]. | Ideal for accelerating BH searches where high accuracy is required but pure DFT is too slow. |
The basin-hopping algorithm is a powerful global optimization technique designed to locate the global minimum on complex potential energy surfaces (PESs), which is crucial for predicting stable molecular and material structures. This method is particularly valuable in computational chemistry and materials science, where it helps researchers identify low-energy configurations of systems ranging from atomic clusters to complex biomolecules and crystalline materials. By transforming the original PES into a collection of interpenetrating staircases, where each plateau corresponds to a local minimum, basin-hopping enables efficient navigation through the energy landscape, overcoming large barriers that would trap simpler minimization methods [4].
Within the context of thesis research on local minima identification, this case study examines the application of basin-hopping to two distinct systems: Lennard-Jones clusters, which serve as benchmark systems in computational physics, and silicon dioxide (SiO₂) materials, which present more complex optimization challenges due to their flexible network structures. The algorithm's performance, limitations, and practical implementation considerations will be explored through troubleshooting guides and experimental protocols relevant to researchers and drug development professionals working with molecular structure prediction [45] [2].
Troubleshooting Guides & FAQs
FAQ: What is the basin-hopping algorithm and how does it work?
Basin-hopping is a stochastic global optimization algorithm that combines random perturbation of coordinates with local minimization. Each iteration consists of:
- Random displacement: Perturbing the current coordinates
- Local minimization: Refining the perturbed structure to the nearest local minimum
- Acceptance test: Deciding whether to accept the new minimum based on the Metropolis criterion
This process effectively transforms the potential energy surface, making it easier to escape local minima and explore different regions of the configuration space. The algorithm is particularly effective for "funnel-like, but rugged" energy landscapes common in molecular and material systems [10] [4].
Common Issue: Algorithm Trapped in Local Minima
Problem: The basin-hopping search repeatedly returns the same local minimum instead of finding the global minimum.
Solutions:
- Increase temperature parameter: Higher T values in the Metropolis criterion allow more uphill moves to escape deep local minima. For SiO₂ systems, temperatures comparable to the typical energy difference between local minima (often 1-100 K in reduced units) are effective [45] [10].
- Adjust step size: The step size should be comparable to the typical separation between local minima. For atomic systems, this often ranges from 0.1-1.0 Å. Implement adaptive step size control using the
target_accept_rate(default 0.5) andstepwise_factor(default 0.9) parameters [10]. - Use structure-based guiding: Instead of energy-based guidance alone, employ structural order parameters. For SiO₂, maximizing the shortest silicon-silicon distance proved more effective than energy guidance alone [45].
- Implement machine learning augmentation: Apply similarity indices and hierarchical clustering to identify unexplored regions of the PES and guide the search toward them [4].
Common Issue: Handling Constraints and Boundaries
Problem: The optimization violates physical constraints or fails to maintain boundary conditions.
Solutions:
- Custom step function: Implement a bounded step function like
RandomDisplacementBoundsto ensure new coordinates stay within physical bounds, especially for portfolio optimization or confined systems [46]. - Constraint functions: Use the
accept_testparameter to define custom acceptance criteria that reject structures violating constraints. For example, in portfolio optimization, this can ensure weights sum to 1 [46] [10]. - Penalty functions: Modify the objective function to include penalty terms for constraint violations rather than relying on the algorithm's built-in constraint handling.
Common Issue: Poor Performance with Large Systems
Problem: Computational efficiency decreases significantly with system size (e.g., SiO₂ 72-atom cells vs. 18-atom cells).
Solutions:
- System-specific step algorithms: For biomolecules, focus on dihedral angle perturbations rather than Cartesian coordinates. For SiO₂ systems, prioritize oxygen bridge manipulations [45] [4].
- Hybrid methods: Combine basin-hopping with other techniques. For protein structure prediction, integrate with umbrella sampling or use mutational basin-hopping for biomolecules [2] [4].
- Parallelization: Utilize the inherent parallelism in basin-hopping by running multiple explorers simultaneously, as implemented in automated PES exploration packages [47].
- Reduced representation: Employ coarse-grained models where appropriate, such as the 4-letter amino acid code in protein folding studies, to decrease computational cost [2].
FAQ: How do I choose appropriate parameters for my system?
Key Parameter Guidelines:
Table: Basin-Hopping Parameter Selection Guide
| Parameter | Recommended Values | System-Specific Considerations |
|---|---|---|
| Temperature (T) | 1-100 (reduced units) | Should be comparable to typical energy difference between local minima; higher for more rugged landscapes |
| Step Size | 0.1-1.0 Å (atomic systems) | Should be comparable to typical separation between minima; adjust based on acceptance rate |
| Iterations (niter) | 100-1000+ | Larger for more complex systems; use niter_success for early termination |
| Minimizer Method | L-BFGS-B, Powell, BFGS | L-BFGS-B for systems with bounds; Powell for derivatives-free optimization |
These parameters should be calibrated for specific systems: for Lennard-Jones clusters, start with T=10-100 and stepsize=0.5; for SiO₂, use structure-based guiding with T=1-10; for portfolio optimization, implement strict boundary control [45] [46] [10].
Experimental Protocols
Protocol 1: SiO₂ Structure Prediction Using GHA and Basin-Hopping
Objective: Locate the global minimum (α-quartz) and low-energy polymorphs of SiO₂ using a hybrid genetic-basin-hopping approach.
Methodology:
- System Setup:
- Implement the Genetic Hybrid Algorithm (GHA) platform with LAMMPS integration for energy evaluation
- Use Tersoff potential with parameters from [20] for SiO₂ interactions
- Define unit cells with 18, 36, or 72 atoms, fixing volume dimensions for interface studies
Basin-Hopping Configuration:
- Apply structure-based guiding instead of energy-guided search
- Implement two guiding metrics: (1) maximize shortest Si-Si distance, (2) maximize calculated order parameter
- Generate multiple basin hopping jump options with weighted selection based on structural properties
Optimization Procedure:
- Execute runs through the GHA preprocessor to generate initial population
- Apply iterative perturbation-minimization-acceptance cycles
- Identify and characterize false attractors (low-energy structures that trap the search)
- Compare final structures with known α-quartz configuration [45]
Key Findings:
- Energy-guided basin-hopping proved detrimental for SiO₂; structure-based guiding more reliable
- Three low-energy structures near the global minimum act as false attractors
- 18-atom cells converge more readily than 36 or 72-atom systems
- Successful location of α-quartz global minimum with appropriate guiding [45]
Protocol 2: Protein Structure Prediction with AMH Potential
Objective: Identify low-energy protein conformations using the Associative Memory Hamiltonian and basin-hopping.
Methodology:
- Coarse-Grained Modeling:
- Implement the AMH/AMC potential with reduced amino acid representation (4 types instead of 20)
- Represent each residue with Cα, Cβ, and O atoms (except glycine)
- Define energy units through native state energy normalization: ε = |EamcN|/4N, where N is residue count
Basin-Hopping Implementation:
- Use random Cartesian coordinate perturbations followed by minimization
- Apply Metropolis acceptance criterion with temperature TAMC = kBT/ε
- For large systems, implement umbrella sampling with basin-hopping to confirm global minima
Performance Optimization:
- Compare with molecular dynamics with simulated annealing
- Employ Gō-model smoothing to reduce frustration from favorable non-native contacts
- Use order parameter Q (structural similarity metric) to track progress [2]
Key Findings:
- Basin-hopping located lower minima and conformations closer to experimental structures than molecular dynamics
- Efficiency decreased for large systems with Cartesian coordinate perturbations
- Bioinformatic techniques successfully reduced energy surface roughness
- Excluded volume improvements guided by basin-hopping produced better structures [2]
Results & Data Analysis
Table: Performance Comparison of Basin-Hopping Applications
| System | Algorithm Variant | Key Metrics | Results |
|---|---|---|---|
| SiO₂ (18-atom) | GHA + Structure-guided BH | Success rate finding α-quartz | Significant improvement over energy-guided BH |
| SiO₂ (36,72-atom) | GHA + Structure-guided BH | Convergence time, energy variance | Longer convergence, tighter energy grouping, more structural variety |
| Protein Folding (AMH) | Cartesian BH | Minimum energy vs experimental | Lower minima and closer to experimental than molecular dynamics |
| Protonated Serine Dimer | ML-augmented BH | Spectral assignment accuracy | Successful interpretation of IRMPD spectrum |
| Alanine Tripeptide | ML-augmented BH | Collision cross-section prediction | Accurate temperature-dependent CCS values |
| Lennard-Jones Clusters | Standard BH | Atoms located in global minimum | Successful for clusters up to 110 atoms [46] |
Table: Computational Settings for Different System Types
| System Type | Recommended Local Minimizer | Temperature Setting | Step Size Control | Constraint Handling |
|---|---|---|---|---|
| Atomic/Material (SiO₂) | L-BFGS-B | T=1-10 | Adaptive with structure-based guiding | Fixed volume dimensions |
| Molecular Clusters | BFGS | T=10-100 | 0.5 Å default | Distance constraints |
| Biomolecules | Powell | T=300K (physical temp) | Dihedral angle focus | Excluded volume constraints |
| Portfolio Optimization | L-BFGS-B | T=1 | Bounded displacement | Weight sum constraint |
The Scientist's Toolkit
Table: Essential Research Reagent Solutions for Basin-Hopping Studies
| Tool/Code | Application Context | Function | Implementation Notes |
|---|---|---|---|
| GHA Platform | SiO₂ structure prediction | Genetic algorithm framework integrated with basin-hopping | Custom implementation with LAMMPS library for energy evaluation |
| AMS PESExploration | General molecular systems | Automated potential energy surface exploration | Commercial software with multiple job types (ProcessSearch, BasinHopping, SaddleSearch) |
| Custom Similarity Functions | Machine learning augmentation | Quantify structural similarity between conformers | Enables hierarchical clustering and multidimensional scaling of PES |
| RandomDisplacementBounds | Constrained optimization | Ensure trial moves respect boundary conditions | Essential for portfolio optimization and confined systems [46] |
| Tersoff Potential | SiO₂ and silicon systems | Bond-order potential for energy evaluation | Fast computation with reasonable accuracy for SiO₂ bonding [45] |
| Associative Memory Hamiltonian | Protein structure prediction | Coarse-grained potential incorporating bioinformatic data | Combines physical models with database information from threading [2] |
Workflow Visualization
Basin-Hopping Algorithm Flowchart
Structure-Based Guiding for SiO₂ Optimization
Key Insights for Practitioners
Successful implementation of basin-hopping for structure prediction requires careful attention to system-specific considerations. For Lennard-Jones clusters, the algorithm has proven exceptionally effective, locating global minima for systems containing up to 110 atoms. However, for more complex systems like SiO₂, standard implementations often require modification, such as the structure-based guiding approach developed in the GHA platform. Biomolecular systems present additional challenges, where machine learning augmentation through similarity indices and hierarchical clustering significantly improves performance.
The temperature parameter T remains one of the most critical factors in basin-hopping success. For systems with clearly defined funnels, lower temperatures (T=1-10) promote convergence to the global minimum, while more rugged landscapes require higher temperatures (T=10-100) to escape deep local minima. Practitioners should implement systematic parameter optimization and consider hybrid approaches that combine basin-hopping with other global optimization techniques for challenging systems [45] [10] [4].
When applying these methods to drug development contexts, particularly for protein-ligand docking or conformer generation, the integration of basin-hopping with experimental validation through spectroscopy or ion mobility becomes crucial. The case studies presented demonstrate that while basin-hopping alone cannot guarantee location of the true global minimum, its combination with experimental data and machine learning approaches provides a powerful framework for structural prediction across diverse scientific domains.
Troubleshooting Guides
Poor Scaling of Parallel Performance
Problem: The overall speedup of the parallel Basin-Hpping (BH) run is less than expected, failing to achieve near-linear reduction in wall-clock time.
Diagnosis: This is commonly caused by synchronization overhead and load imbalance between parallel processes. Performance gains typically plateau as the number of concurrent evaluations increases. One study observed that "near-linear speedup for up to eight concurrent local minimizations" is possible, but "beyond which efficiency gains become modest due to synchronization overhead." [9]
Solution:
- Optimize Core Count: Limit the number of concurrent local searches to 8 or fewer for a single BH step to minimize overhead. [9]
- Profile Workload: Ensure the computational cost of each local minimization is roughly equal to avoid situations where one slow process blocks the entire iteration.
- Check System Resources: Monitor CPU and memory usage to confirm that the system is not being over-subscribed, which can lead to resource contention.
Algorithm Trapped in Local Minima
Problem: The parallel BH search appears kinetically trapped, repeatedly finding the same sub-optimal local minimum despite the increased sampling.
Diagnosis: The "temperature" in the Metropolis acceptance criterion may be set too low, or the perturbation stepsize is insufficient to push the system into a new basin of attraction. Furthermore, increasing the number of concurrent trials per step does not fundamentally change the algorithm's ability to escape very deep local minima. [4] [12]
Solution:
- Implement Adaptive Step-Size: Use an algorithm that dynamically adjusts the perturbation step size based on the recent acceptance rate, aiming for a target (e.g., 50%) to balance exploration and refinement. [48] [9]
- Adjust Metropolis Temperature: Increase the "temperature" parameter to allow for a higher probability of accepting uphill moves, facilitating escape from local minima. [4] [12]
- Combine with Machine Learning: Augment the search with unsupervised machine learning techniques. By calculating similarity indices and using hierarchical clustering on collected structures, you can identify unexplored regions of the potential energy surface and guide perturbations away from already-sampled minima. [4]
Inconsistent Results Across Parallel Runs
Problem: Multiple independent parallel BH runs on the same system yield different final "best" structures, reducing confidence in the identified global minimum.
Diagnosis: BH is an inherently stochastic algorithm, and "identifying the global minimum via a finite, stochastic search is not guaranteed." This is a fundamental characteristic of the method, and confidence is built through multiple independent runs. [4]
Solution:
- Increase Number of Iterations: Run each parallel BH simulation for more iterations (e.g., thousands instead of hundreds) to improve the exhaustiveness of each search. [12]
- Perform Ensemble Runs: Execute several completely independent parallel BH runs from different random starting points and compare the results. Consensus across multiple runs increases confidence. [4]
- Validate with Experiments: Where possible, use experimental data (e.g., from spectroscopy or ion mobility) to validate and rationalize the collection of low-energy structures found. [4]
Frequently Asked Questions (FAQs)
Q1: What is the core idea behind parallelizing the Basin-Hopping algorithm?
A1: The standard BH algorithm cycles through perturbing a structure, locally minimizing it, and then accepting or rejecting it. The most effective parallelization strategy is, at each BH step, to generate multiple perturbed trial structures and then distribute their local minimizations across available CPU cores. The best result from these concurrent local searches is then used in the Metropolis acceptance test, improving the quality of the step and reducing wall-clock time. [9] [49]
Q2: Can I parallelize the scipy.optimize.basinhopping function directly?
A2: Parallelizing the core algorithm of scipy.optimize.basinhopping is not straightforward, as the implementation is not designed for it. The slowness often comes from the user-defined objective function. If the function itself is slow, you can try to parallelize its internal computations. Alternatively, consider using a custom implementation of parallel BH, or explore other global optimizers in SciPy like differential_evolution or shgo that natively support parallel function evaluations. [50]
Q3: How do I choose the number of parallel trials per Basin-Hopping step?
A3: The optimal number is system-dependent. Start with a value between 4 and 8, as research has shown this range often provides a "nearly linear speedup". Using more than 8 concurrent evaluations can lead to diminishing returns due to synchronization overhead. The goal is to balance deeper exploration of the neighborhood at each step with computational efficiency. [9] [49]
Q4: What local minimizer should I use for the parallel searches?
A4: Gradient-based methods are typically preferred for efficiency. The L-BFGS-B algorithm is a common and robust choice for this task, and it is the default in many implementations, including the one described in the referenced research. [9] [12] The ASE optimization library also offers various reliable local optimizers like BFGS and FIRE for atomic systems. [43]
Performance Data and Configuration
Quantitative Performance of Parallel Basin-Hopping
The following table summarizes key quantitative findings on the efficiency of parallel Basin-Hopping implementations, as reported in the literature.
Table 1: Performance Metrics of Parallel Basin-Hopping
| Metric | Reported Value / Finding | Context / Conditions | Source |
|---|---|---|---|
| Computational Speedup | Nearly linear speedup | Achieved for up to 8 concurrent local minimizations. | [9] |
| Performance Scaling | Diminishing returns | Observed when using more than 8 parallel candidates due to synchronization overhead. | [9] |
| Convergence Improvement | Increased success rate & speed | Using multiple parallel local searches per step helps overcome traps in hard optimization problems. | [49] |
| Typical Iterations | 200 to 10,000+ | The number of BH steps required depends on the complexity of the potential energy surface. | [12] |
Experimental Protocol: Parallel BH for Lennard-Jones Clusters
This protocol is adapted from recent research on accelerating BH for atomic clusters. [48] [9]
1. Initialization:
- Objective Function: Define the total energy of the system using the Lennard-Jones potential: ( E{LJ} = 4\epsilon \sum{i
- Initial Structure: Provide an initial atomic geometry, for example, from a
.moldenfile containing Cartesian coordinates. - Parameters: Set the initial step size for perturbations (e.g., 0.5), the number of parallel trials per step
n(e.g., 8), and the Metropolis temperature (e.g., ( T = 1.0 )).
2. Core Iterative Loop: For each step in the total number of iterations (e.g., 200 steps):
- Perturbation: Generate
nnew trial structures by applying random displacements to the current best structure. The maximum displacement is controlled by thestepsizeparameter. - Parallel Local Optimization: Distribute the
ntrial structures to a multiprocessing pool (e.g., usingmultiprocessing.Poolin Python). Perform a local minimization on each structure using the L-BFGS-B algorithm. - Selection: From the
nminimized candidates, select the one with the lowest energy. - Metropolis Criterion: Accept or reject the selected candidate based on its energy relative to the current state. If the new energy is lower, accept it. If it is higher, accept it with a probability of ( \exp(-\Delta E / T) ), where ( \Delta E ) is the energy difference.
- Adaptation (Optional): Every 10 steps, calculate the recent acceptance rate. Adjust the
stepsizeto maintain a target acceptance rate of ~50%.
3. Output and Analysis:
- The algorithm outputs a file (e.g.,
input_accepted.molden) containing all accepted structures during the search. - Analyze the trajectory of accepted energies and the final low-energy structures to identify the global minimum and key local minima.
Workflow Diagram
The diagram below illustrates the logical flow and parallelization strategy of the accelerated Basin-Hopping algorithm.
The Scientist's Toolkit: Research Reagents & Solutions
Table 2: Essential Computational Tools for Parallel Basin-Hopping Research
| Tool / Resource | Function / Purpose | Application Example |
|---|---|---|
| Lennard-Jones Potential | A classic model potential for pairwise atomic interactions; serves as a benchmark for testing optimization algorithms. | Prototypical system for evaluating the performance of parallel BH on atomic clusters. [48] [9] |
| L-BFGS-B Optimizer | A local minimization algorithm that is efficient and well-suited for use within the inner loop of BH. | The default local minimizer in the described parallel BH implementation for relaxing perturbed structures. [9] [12] |
| Python Multiprocessing | A standard library in Python for spawning processes to leverage multiple CPU cores. | Used to create a pool of workers for conducting concurrent local energy minimizations. [9] |
| ASE (Atomic Simulation Environment) | A set of tools and Python modules for setting up, manipulating, and analyzing atomistic simulations. | Provides various local optimizers (BFGS, FIRE) and calculators for running BH on material systems. [43] |
| Similarity Index & Clustering | Concepts from unsupervised machine learning used to compare and group molecular geometries. | Augments BH by identifying unique minima and guiding the search towards unexplored regions of the energy landscape. [4] |
| 2-HP-β-Cyclodextrin | A solubility-enhancing agent used in drug formulation studies. | Served as a case study where in-silico BH was used to propose putative binding modes for inclusion complexes with drug ligands. [51] |
Advanced Strategies: Enhancing Basin-Hopping Efficiency and Overcoming Common Pitfalls
Adaptive Step-Size Control for Dynamic Exploration-Exploitation Balance
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: My basin-hopping simulation is converging too quickly to local minima and missing the global minimum. How can I improve its exploration capability?
This is a classic sign of insufficient exploration. The adaptive step-size is likely too small, trapping the algorithm in a suboptimal region.
- Solution: Increase the target acceptance rate for the adaptive controller from 0.5 to a higher value, such as 0.6 or 0.7. This encourages the algorithm to accept more uphill moves, facilitating escape from local minima [9]. Simultaneously, ensure your step-size adjustment magnitude is sufficient; a very small adjustment gain will take too long to react. You can also temporarily increase the number of parallel trial structures evaluated at each step to sample the energy landscape more broadly [9].
Q2: My simulation is oscillating wildly and failing to converge, even to a local minimum. What is happening?
This behavior indicates excessive exploration and a failure to exploit promising regions. The step-size for perturbations is likely too large.
- Solution: Reduce the target acceptance rate for the adaptive controller, for example, to 0.3 or 0.4. This makes the Metropolis criterion more stringent, accepting fewer uphill moves and promoting refinement within a basin [9]. Verify that the formula for dynamic step-size adjustment is correctly implemented. A check of the step-size's evolution over time can confirm it is decreasing appropriately as the run progresses.
Q3: How do I know if my adaptive step-size controller is working correctly?
Monitoring specific metrics is key to diagnosing the controller's performance.
- Diagnosis: Log the step-size and the instantaneous acceptance rate at regular intervals (e.g., every 10-20 steps) [9]. Plot these values against the number of iterations. You should observe the step-size dynamically adjusting in response to the acceptance rate. If the acceptance rate is consistently above the target, the step-size should increase, and vice-versa. A flat step-size graph suggests a malfunctioning adaptation mechanism.
Q4: What is the difference between exploration and exploitation in the context of the basin-hopping algorithm?
This is a fundamental trade-off in optimization [52].
- Exploration involves making larger perturbations to the current structure or accepting higher-energy states to escape local minima and probe new regions of the potential energy surface (PES). This is riskier but is necessary for locating the global minimum [53].
- Exploitation involves making smaller perturbations and focusing on local minimization to deeply search the vicinity of a promising minimum. This refines a solution but can lead to entrapment in a local minimum [53]. The adaptive step-size directly manages this balance: a larger step promotes exploration, while a smaller step favors exploitation [9] [54].
Troubleshooting Common Experimental Issues
Table 1: Common Issues and Corrective Actions in Basin-Hopping Simulations
| Problem | Possible Causes | Diagnostic Checks | Corrective Actions |
|---|---|---|---|
| Premature Convergence | Step-size too small; Target acceptance rate too low [9]. | Plot energy vs. iteration; Check final step-size value. | Increase target acceptance rate; Increase initial step-size. |
| Erratic, Non-Converging Behavior | Step-size too large; Ineffective local minimizer [9]. | Check acceptance rate history (should be very high). | Reduce target acceptance rate; Verify local optimizer convergence. |
| Slow Performance | High cost of energy/force calculations; Too many parallel trials. | Profile code to identify bottleneck. | Use a more efficient potential; Optimize number of parallel workers [9]. |
| Algorithm Ignoring Adaptive Control | Bug in adaptation logic; Adaptation interval too long. | Log step-size every iteration to see if it changes. | Check the "if" statement for step-size update; Reduce adaptation frequency. |
Experimental Protocols and Methodologies
Protocol 1: Implementing an Adaptive Step-Size Controller
This protocol outlines the integration of an adaptive step-size mechanism into a standard basin-hopping algorithm to dynamically balance exploration and exploitation [9] [55].
- Initialization: Define an initial step-size (e.g., 0.5 Å), a target acceptance rate (e.g., 0.5), and an adaptation interval (e.g., every 10 accepted steps) [9].
- Basin-Hopping Cycle: For each iteration
N: a. Perturbation: Apply a random displacement to the current atomic coordinates, where the maximum displacement is controlled by the current step-size. b. Local Minimization: Use a local optimizer (e.g., L-BFGS-B) to quench the perturbed structure to the nearest local minimum [9]. c. Metropolis Criterion: Accept or reject the new minimum based on its energy and a Boltzmann probability at a fixed temperature (e.g., T=1.0) [9]. - Adaptation Step: Every
Kaccepted steps (whereKis the adaptation interval): a. Calculate the empirical acceptance rate over the most recentKsteps. b. Adjust Step-size: If the acceptance rate is above the target, increase the step-size by a small factor (e.g., 5%). If it is below, decrease the step-size. Implement bounds to prevent the step-size from becoming excessively large or small [9]. - Termination: Continue until a convergence criterion is met (e.g., maximum iterations, no improvement for a set number of steps).
The logical flow of this adaptive control mechanism is shown below.
Protocol 2: Benchmarking on a Known System (Lennard-Jones Clusters)
To validate the performance of your adaptive basin-hopping implementation, test it on a well-studied system like the Lennard-Jones (LJ) cluster, a standard benchmark in global optimization studies [9].
- System Selection: Choose a cluster known for a complex energy landscape, such as LJ₃₈, which has a famous "funneled" landscape that is difficult to navigate [9].
- Potential Setup: Implement the Lennard-Jones potential:
E_LJ = 4ε Σ [(σ/r_ij)¹² - (σ/r_ij)⁶], with ε=σ=1 in reduced units [9]. - Comparative Experiment: a. Run the basin-hopping algorithm with a fixed step-size. b. Run the basin-hopping algorithm with the adaptive step-size controller from Protocol 1.
- Performance Metrics: For each run, record: a. The lowest energy minimum found. b. The number of iterations to find the global minimum. c. The evolution of the step-size (for the adaptive run). d. The final acceptance rate.
- Analysis: Compare the success rate and efficiency of both methods in locating the known global minimum of the benchmark system. The adaptive method should demonstrate a more robust and efficient search [9].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for Basin-Hopping Research
| Item | Function / Explanation | Example / Note |
|---|---|---|
| L-BFGS-B Optimizer | A quasi-Newton local minimization algorithm efficient for problems with many variables [9]. | Found in scipy.optimize. Ideal for relaxing perturbed atomic structures. |
| Lennard-Jones Potential | A classic pair potential used for rapid testing and benchmarking of optimization algorithms [9]. | Its known global minima for clusters make it an excellent validation tool. |
| Metropolis Criterion | A probabilistic rule to decide whether to accept a higher-energy solution, allowing escape from local minima [9]. | Uses a temperature parameter T to control the probability of acceptance. |
| Multiprocessing Pool | A programming construct for parallel evaluation of multiple trial structures, drastically reducing wall-clock time [9]. | Python's multiprocessing.Pool can be used to parallelize local minimizations. |
| Molden File Format | A common format for storing molecular coordinates and other data, useful for input and output of structures [9]. | Allows visualization of accepted structures and reaction pathways. |
The overall workflow of an adaptive basin-hopping experiment, integrating the components from the toolkit, is visualized below.
The Basin-hopping algorithm has established itself as a powerful global optimization method, particularly effective for navigating the complex energy landscapes common in chemical physics, materials science, and drug development. By combining random perturbation of coordinates with local minimization, this algorithm efficiently maps potential energy surfaces into basins of attraction, enabling researchers to identify low-energy configurations of atomic clusters, biological macromolecules, and complex material systems [2] [9]. Despite its sophisticated approach, the algorithm's practical implementation presents specific challenges related to parameter selection, constraint handling, and convergence verification that can significantly impact research outcomes.
This technical support center addresses the most frequent operational challenges encountered when implementing Basin-hopping methodologies, providing researchers with practical troubleshooting guidance framed within the broader context of local minima identification research. The following sections synthesize current implementations across computational chemistry and materials science domains to deliver actionable protocols for optimizing algorithmic performance.
Core Concepts: Understanding the Basin-Hopping Algorithm
What is the fundamental mechanism behind Basin-hopping's ability to escape local minima?
Basin-hopping employs a two-phase cycle that transforms the original potential energy surface: (1) random perturbation of current coordinates followed by (2) local minimization of the resulting structure. This approach effectively removes high energetic barriers by mapping the landscape into a collection of basins of attraction, enabling more efficient transition between low-energy regions. The algorithm accepts or rejects new minima based on the Metropolis criterion, which incorporates a "temperature" parameter (T) to control the probability of accepting higher-energy configurations [2] [3]. This combination of coarse-grained landscape exploration with local refinement allows the algorithm to escape kinetic traps that would immobilize simpler optimization methods.
How does Basin-hopping differ from molecular dynamics with simulated annealing?
Unlike molecular dynamics with simulated annealing, which relies on thermal fluctuations to overcome barriers, Basin-hopping uses gradient-based local minimization after each perturbation. Research comparing both approaches for protein structure prediction found that Basin-hopping succeeded in locating both lower minima and conformations closer to experimental structures, particularly for small systems. This advantage stems from Basin-hopping's ability to avoid the slow kinetics of glass-like transitions that can hinder simulated annealing approaches [2].
Frequently Asked Questions (FAQs)
FAQ 1: What criteria should guide my selection of the temperature parameter (T) for the Metropolis acceptance criterion?
The temperature parameter T should be comparable to the typical energy difference between local minima in your system, not the height of barriers between them. For best results, T must be calibrated to your specific energy landscape:
- Optimal Range: The SciPy documentation recommends that T "should be comparable to the separation (in function value) between local minima" [3].
- Acceptance Rate Monitoring: Implement adaptive schemes that monitor acceptance rates and adjust T to maintain optimal exploration versus exploitation balance.
- System-Specific Considerations: For protein folding applications using associative memory Hamiltonians, reduced temperature units (TAMC = kBT/ϵ) are employed, where ϵ is defined in terms of the native state energy [2].
FAQ 2: How can I determine the optimal step size for coordinate perturbations in my system?
Step size selection is crucial and system-dependent. The optimal value should be comparable to the typical separation between local minima in argument values:
- Default Values: The SciPy implementation defaults to stepsize=0.5, but this requires adjustment for specific applications [3].
- Adaptive Approaches: Modern implementations dynamically adjust step size based on recent acceptance rates. One Python implementation recalculates stepsize every 10 steps to approach a target acceptance rate of 50% using a stepwise factor of 0.9 [9].
- System-Specific Examples: In SiO₂ optimization, step size must be tuned relative to unit cell dimensions, while for Lennard-Jones clusters, step size relates to the characteristic atomic spacings [45].
FAQ 3: Why does the algorithm appear to ignore constraints during optimization?
Constraints may seem ignored because Basin-hopping applies them only during the local minimization phase, not the initial random perturbation:
- Algorithmic Structure: Random jumps occur before constraints are enforced during local minimization, potentially generating trial structures outside feasible regions [19].
- Solution Strategies: Implement a custom
take_steproutine that incorporates constraint information directly into the perturbation phase, or use aaccept_testfunction to immediately reject constraint-violating configurations [3] [19]. - Method Selection: When using COBYLA as the local minimizer, ensure constraints are properly formatted as inequality dictionaries, but recognize that initial perturbations may still temporarily violate constraints [19].
FAQ 4: What strategies can prevent prolonged trapping in persistent local minima?
Persistent trapping indicates insufficient exploration, which can be addressed through:
- Population-Based Approaches: Implement parallel Basin-hopping variants that evaluate multiple trial structures simultaneously, significantly reducing trapping probability [9] [24].
- Alternative Guiding Metrics: For challenging systems like SiO₂, replace energy-based guidance with structure-based metrics such as maximizing the shortest silicon-silicon bond distance or optimizing order parameters [45].
- Advanced Algorithm Variants: Incorporate multicanonical Monte Carlo sampling within the Basin-hopping framework, which actively encourages escape from deep energy traps [56].
- Step Size Adjustment: Increase exploration by dynamically modifying step size when trapping is detected through low acceptance rates over multiple iterations [9].
FAQ 5: How can I verify that the identified minimum is truly global rather than another low-lying local minimum?
Verification remains challenging for stochastic global optimization, but these strategies improve confidence:
- Multiple Random Restarts: Execute the algorithm from diverse initial configurations to assess convergence consistency [3].
- Statistical Validation: For atomic clusters, compare results against known databases of optimized structures, such as the Cambridge Cluster Database [3].
- Umbrella Sampling Integration: Implement umbrella sampling with Basin-hopping using structure-constraining potentials to systematically explore order parameters and validate comprehensive sampling [2].
- Cross-Method Validation: Compare results with those obtained from genetic algorithms or other global optimization methods [57].
Troubleshooting Guides
Problem 1: Poor Acceptance Rate and Insufficient Landscape Exploration
Symptoms
- The algorithm consistently rejects proposed moves
- Limited diversity in sampled minima
- Inability to locate known global minima from different starting points
Diagnosis and Resolution
Table 1: Troubleshooting Poor Acceptance Rates
| Symptom | Potential Cause | Solution | Implementation Example |
|---|---|---|---|
| Consistently low acceptance rate | Step size too large for energy landscape | Implement adaptive step size control | Adjust step size every 10 iterations based on acceptance rate [9] |
| Trapping in specific energy basins | Temperature parameter too low | Increase T to accept more uphill moves | Set T comparable to typical energy differences between local minima [3] |
| Limited structural diversity | Inadequate perturbation magnitude | Modify take_step routine for system-specific moves | For atomic clusters, combine Cartesian displacements with rotation moves [9] |
| Persistent localization | Homogeneous perturbation strategy | Implement parallel candidate evaluation | Generate and minimize multiple trial structures simultaneously [9] [24] |
Protocol: Implement adaptive step size control with the following Python pseudocode:
Problem 2: Inefficient Convergence for High-Dimensional Systems
Symptoms
- Exponential increase in computation time with system size
- Excessive function evaluations required per minimization
- Incomplete sampling of configuration space
Diagnosis and Resolution
Table 2: Optimization Strategies for Large Systems
| Challenge | Impact | Mitigation Strategy | Research Support |
|---|---|---|---|
| Computational cost of local minimization | Limits system size and sampling | Implement parallel local minimization | Parallel evaluation of multiple trial structures [9] [24] |
| Rugged energy landscapes | Slows convergence and traps searches | Incorporate smoothing potentials or bioinformatic guidance | Use associative memory Hamiltonians with optimized frustration [2] |
| Increasing local minima count | Reduces probability of finding global minimum | Hybrid algorithms combining BH with genetic approaches | Genetic Hybrid Algorithm (GHA) platform [45] |
| Parameter sensitivity | Requires extensive tuning for each system | Adaptive parameter control schemes | Dynamic adjustment of step sizes and temperatures [9] [3] |
Protocol: For parallel evaluation of candidate structures:
Experimental Protocols and Methodologies
Standard Basin-Hopping Implementation
The core Basin-hopping algorithm follows this established workflow:
- Initialization: Define initial coordinates x₀, temperature T, step size, and local minimizer parameters [3]
- Iteration Cycle:
- Perturbation: Apply random displacement to all coordinates (default: uniform distribution within ± step size)
- Local Minimization: Execute local optimization from perturbed coordinates using specified method (e.g., L-BFGS-B, BFGS)
- Acceptance Test: Evaluate candidate using Metropolis criterion: accept if f(new) < f(old) or with probability exp(-(f(new)-f(old))/T)
- Termination: Continue for specified iterations or until convergence criteria satisfied (e.g., niter_success with no improvement)
Advanced Implementation: Parallel Basin-Hopping with Adaptive Step Size
Recent implementations enhance efficiency through parallelism and adaptive control:
Parallel Candidate Evaluation:
- Generate multiple trial structures (typically 4-8) through independent perturbations
- Execute concurrent local minimizations using multiprocessing or distributed computing
- Select the lowest-energy structure from the minimized candidates for acceptance testing [9]
Adaptive Step Size Control:
Termination Criteria Enhancement:
- Implement niter_success to stop after specified iterations without improvement
- Combine with maximum iteration count as fallback termination
Table 3: Key Computational Tools for Basin-Hopping Research
| Tool/Resource | Function | Application Context |
|---|---|---|
| SciPy optimize.basinhopping [3] | Reference implementation with adaptive features | General-purpose optimization, educational use |
| LAMMPS [45] | Molecular dynamics with energy/force evaluation | Materials science, SiO₂ optimization, interface structures |
| Custom Python implementation with parallel processing [9] | Specialized for atomic cluster optimization | Lennard-Jones clusters, benchmark studies |
| Genetic Hybrid Algorithm (GHA) platform [45] | Hybrid optimization framework combining BH with genetic algorithms | Complex materials, oxide thin films |
| Cambridge Cluster Database [3] | Repository of known global minima structures | Validation and benchmarking for atomic clusters |
| Associative Memory Hamiltonian [2] | Bioinformatically-informed potential energy function | Protein structure prediction, mildly frustrated landscapes |
Performance Optimization and Validation Techniques
Efficiency Comparison of Enhancement Strategies
Table 4: Performance Comparison of Basin-Hopping Enhancements
| Method | Advantages | Limitations | Reported Efficiency Gains |
|---|---|---|---|
| Standard Basin-Hopping [3] | Simple implementation, generally applicable | Susceptible to trapping in complex landscapes | Baseline for comparison |
| Parallel candidate evaluation [9] | Near-linear speedup, reduced trapping | Synchronization overhead with many processors | 8x speedup with 8 concurrent minimizations |
| Multicanonical Basin-Hopping [56] | Enhanced escape from deep minima | Increased implementation complexity | Superior efficiency for LJ150-185 clusters |
| Population-based variants (BHPOP) [24] | Improved robustness, better exploration | Higher computational cost per iteration | Competitive with CMA-ES on benchmark functions |
| Structure-based guidance [45] | Effective for specific material systems | Reduced transferability | Successful for SiO₂ global minimum identification |
Validation Methodologies
Statistical Validation Protocol:
- Execute multiple independent runs from different initial configurations
- Compare obtained minima for consistency across runs
- Compute success probability as fraction of runs finding putative global minimum
- For atomic systems, compare bond lengths, coordination numbers, and symmetry elements with experimental data where available
Order Parameter Analysis:
- Define system-specific order parameters (e.g., Q in protein folding [2])
- Implement umbrella sampling along order parameter coordinates
- Construct free energy profiles to identify all relevant minima
- Verify that Basin-hopping samples across the entire order parameter range
Augmenting BH with Machine Learning for Intelligent PES Exploration
Frequently Asked Questions (FAQs)
FAQ 1: What is the core principle of the Basin-Hopping (BH) method? The Basin-Hopping (BH) algorithm is a global optimization technique designed to explore complex Potential Energy Surfaces (PES). Its core principle involves transforming the original PES into a collection of interpenetrating staircases. This is achieved by associating any point in configuration space with the local minimum found by starting a local geometry optimization from that point. This transformation effectively removes transition state regions, simplifying the landscape for more efficient global minimum location without altering the identity of the global minimum itself [58].
FAQ 2: How can Machine Learning (ML) specifically enhance the Basin-Hopping method? Machine Learning can augment BH in several key ways. It can act as a surrogate model, providing rapid predictions of molecular energies and forces, which drastically reduces the number of expensive ab initio calculations required during the local minimization steps [59]. Furthermore, ML can guide the global search strategy; for instance, Bayesian Optimization can intelligently tune critical hyperparameters of the BH algorithm, such as step sizes or temperature settings, or even directly propose new, promising candidate structures for the next BH step, thereby improving the overall efficiency of PES exploration [60] [59].
FAQ 3: My BH simulation is trapped in a specific region of the PES and cannot find new minima. What should I do? This is a classic sign of the algorithm being stuck in a deep local energy funnel. You can troubleshoot this by:
- Adjusting the Step Size: The step size for generating new trial configurations is crucial. A step size that is too small will not allow the search to escape the current basin. Use hyperparameter optimization to find an optimal value [61].
- Implementing an Enhanced Algorithm: Consider switching from the standard BH algorithm to an enhanced version like Multicanonical Basin-Hopping [56]. This method combines BH with multicanonical Monte Carlo, which encourages the system to escape energy traps more effectively by using a non-Boltzmann weighting function.
- Leveraging a Surrogate Model: Train an ML model on the structures and energies already computed. Use this model to screen a large number of trial moves, only performing expensive ab initio calculations on the most promising candidates predicted by the ML model to be in a new, low-energy basin [59].
FAQ 4: How do I set up an automated ML-augmented BH workflow for a new molecular system? A robust automated workflow involves several integrated components, as visualized below.
Automated ML-BH Workflow
- Initial Data Generation: Start with a small set of diverse molecular structures and compute their high-fidelity energies and forces using a quantum chemistry package like PySCF [62].
- Model Training: Use this data to train a surrogate ML model (e.g., a deep neural network) to predict energy and forces.
- Integration with BH: The BH algorithm uses this fast ML model for its local minimization steps and to evaluate trial moves.
- Active Learning Loop: New structures discovered by BH are sent back for high-fidelity calculation. Their results are added to the training dataset, which is used to periodically retrain and improve the ML model, creating a closed, self-improving loop [59].
Troubleshooting Guides
Issue 1: Inefficient or Slow Global Exploration
Problem: The BH algorithm is not efficiently locating low-energy minima and is re-sampling known regions of the PES.
Solution: Implement an intelligent sampling strategy using a Monte Carlo-based acquisition function from Bayesian Optimization.
- Detailed Protocol:
- Define the Search Space: Identify the key parameters for your BH run you wish to optimize (e.g., max step size, temperature for Monte Carlo acceptance).
- Choose a Surrogate Model: Model the relationship between BH parameters and performance (e.g., lowest energy found) using a Gaussian Process (GP), as supported by the BoTorch library [60].
- Select an Acquisition Function: Use a Monte Carlo acquisition function, such as qExpectedImprovement (qEI). This function evaluates the potential of multiple candidate points (q>1) simultaneously, which is ideal for parallel computing resources.
- Optimize the Acquisition Function: Maximize the qEI function using gradient-based methods to propose the next, most promising set of BH parameters to evaluate. This step is made efficient in BoTorch through re-parametrization tricks that allow gradient computation [60].
- Iterate: Run BH with the new parameters, observe the outcome (e.g., new lowest energy), update the GP model, and repeat.
The following diagram illustrates the logical relationship between these components and how they guide the BH algorithm.
Intelligent Parameter Guidance Logic
Issue 2: Handling High-Dimensional and Noisy Landscapes
Problem: For systems with many degrees of freedom (e.g., solvated molecules), the PES is high-dimensional and noisy, making it difficult for standard BH to locate the true global minimum.
Solution: Employ a framework like Optuna for robust hyperparameter optimization and integrate enhanced sampling techniques.
- Detailed Protocol:
- Define the Objective Function: Create a function that takes BH hyperparameters (e.g., step size, number of local optimization steps) as input, runs the BH simulation, and returns a scalar objective value (e.g., the predicted energy of the best-found minimum).
- Configure the Optimizer: Initialize an Optuna
studyobject, specifying the minimization direction. Utilize a Tree-structured Parzen Estimator (TPE) sampler for intelligent hyperparameter suggestion [61]. - Incorporate Pruning: Use Optuna's pruning capabilities (e.g.,
MedianPruner) to automatically halt unpromising trials early, saving significant computational resources [61]. - Leverage Multicanonical Sampling: For the local exploration within BH, consider replacing a standard minimizer with a method that incorporates multicanonical sampling. This encourages barrier crossing and can be more effective in complex, trap-filled landscapes [56].
Quantitative Data on Enhanced BH Methods
The following table summarizes key algorithmic variants of BH and their reported performance improvements.
Table 1: Performance Comparison of Basin-Hopping Algorithm Variants
| Algorithm Name | Key Innovation | Reported Efficiency / Performance | Test System |
|---|---|---|---|
| Standard Basin-Hopping [58] | Transformation of PES into staircase of minima | Located lowest known structures for LJ clusters up to 110 atoms | Lennard-Jones (LJ) Clusters |
| Multicanonical Basin-Hopping [56] | Integration with multicanonical Monte Carlo to escape energy traps | More efficient than original BH method | LJ clusters of 150-185 particles |
| Quantum Basin-Hopping [63] | Incorporation of quantum amplitude amplification and gradient estimation | Theoretical acceleration proportional to sqrt(# of basins) and domain dimension | Theoretical Continuous Optimization |
The Scientist's Toolkit
Table 2: Essential Research Reagents & Computational Tools
| Item / Software | Function in ML-Augmented BH | Relevant Context |
|---|---|---|
| BoTorch [60] | Provides Monte Carlo acquisition functions for Bayesian Optimization, enabling gradient-based optimization of complex objectives to guide BH. | Essential for intelligent parameter tuning and parallel candidate suggestion. |
| Optuna [61] | A hyperparameter optimization framework that simplifies the process of defining and optimizing BH parameters with advanced pruning and sampling algorithms. | Crucial for automating the search for optimal BH simulation parameters. |
| PySCF [62] | A quantum chemistry software package used for performing the high-fidelity energy and force calculations required for local minimization and generating training data. | Acts as the source of truth for the PES. |
| geometric [62] | A library for geometry optimization, often used as the local minimizer within the BH algorithm when coupled with PySCF. | Handles the local exploration step in the BH cycle. |
| Deep Neural Network (DNN) | Serves as a surrogate model for the PES, providing fast and accurate predictions of energy and atomic forces, drastically reducing computational cost. | The core ML component for accelerating the search [59]. |
Population-Based Variants (BHPOP) for Improved Robustness
Frequently Asked Questions (FAQs)
Q1: What is BHPOP and how does it fundamentally differ from standard Basin-Hopping? Standard Basin-Hpping (BH) is a trajectory-based algorithm that uses a single candidate solution. In each step, this candidate is perturbed, locally minimized, and then accepted or rejected based on a Monte Carlo criterion [2] [24]. In contrast, BHPOP is a population-based variant. Instead of a single candidate, it maintains and evolves a population of candidate solutions. This allows the algorithm to explore multiple promising regions of the complex energy landscape simultaneously, making it less likely to become trapped in a single deep but local minimum and improving its overall robustness and convergence reliability [24].
Q2: My BHPOP run is not converging to good minima on a complex, high-dimensional function. What could be wrong? Poor convergence in high-dimensional spaces often stems from an improperly tuned strategy for generating new candidate solutions. Below are common issues and their fixes:
- Problem: Ineffective Perturbations. The step size for perturbing population members is either too small (leading to insufficient exploration) or too large (causing chaotic jumps and wasted function evaluations).
- Solution: Implement an adaptive step size. Monitor the acceptance rate of new minima. Aim for an acceptance rate between 20% and 40%. If the rate is too low, reduce the step size; if it's too high, increase it.
- Problem: Loss of Population Diversity. The population has prematurely converged, meaning all individuals are trapped in the same basin of attraction.
- Solution: Introduce a diversity maintenance mechanism. Before adding a new minimum to the population, check its structural similarity (e.g., using root-mean-square deviation or a difference in objective function value) to existing members. If it is too similar to an existing member, reject it unless it has a lower energy, or implement a "crowding" technique to replace similar individuals.
- Problem: Inefficient Local Minimization.
- Solution: For the local search step, choose a minimizer that is appropriate for your problem's characteristics (e.g., L-BFGS for smooth functions, COBYLA for derivative-free problems). Set strict convergence criteria to avoid overly long minimizations on unproductive points.
Q3: How does BHPOP's performance compare to other established metaheuristics? Recent comparative studies have shown that BHPOP is a highly competitive algorithm. The table below summarizes a performance comparison based on benchmark testing [24]:
| Algorithm | Performance Summary | Typical Use Case |
|---|---|---|
| BHPOP | Almost as good as CMA-ES on synthetic benchmark functions and better on hard, real-world energy minimization problems. | Quick and reliable results on an unknown problem, especially complex physical systems. |
| CMA-ES | Often top performer on synthetic benchmark function suites. | Problems where derivative estimation is feasible and high precision is required. |
| Differential Evolution (DE) | Good general performance, but may be outperformed by BHPOP and CMA-ES on highly multimodal problems. | A robust default choice for a wide range of continuous problems. |
| Particle Swarm Optimization (PSO) | Performance can be highly dependent on parameter tuning. | Problems with smooth, continuous landscapes. |
Q4: How do I choose between "significant structures" and "raw structures" Basin-Hopping? This choice relates to how the algorithm's state is maintained after a perturbation and minimization step [6].
- Significant Structures Basin Hopping (SSBH): After a new structure is created (perturbation + minimization), the algorithm's current state is updated to this new, minimized structure. This is the more common and often more effective approach, as it focuses the search on the low-energy "significant" configurations.
- Raw Structures Basin Hopping (RSBH): The algorithm's state remains the unminimized ("raw") structure, even though the energy used for the acceptance criterion is that of its minimized form. This can lead to less efficient exploration.
Recommendation: Prefer the SSBH approach, as research has found it performs better for atomic and molecular clusters when both use optimized step sizes [6]. Implement RSBH only if you have a specific reason to do so.
Troubleshooting Guides
Issue 1: Algorithm Is Not Locating the Global Minimum
| Symptoms | Possible Causes | Diagnostic Steps | Solutions |
|---|---|---|---|
| Consistent convergence to the same, sub-optimal local minimum. | Population diversity is too low. | Track the average pairwise distance (or energy difference) between population members over iterations. | Increase the mutation step size. Introduce a diversity-enforcement mechanism (see FAQ 2). |
| Inadequate sampling of the search space. | Check the number of unique basins (minima) found in the search history. | Increase the population size. Run the algorithm for more generations. | |
| The best-found solution improves very slowly. | The local minimizer is inefficient or failing. | Profile the code to see where most of the computation time is spent. | Switch to a more efficient local minimization algorithm (e.g., L-BFGS). Loosen the tolerance of the local minimizer to save time. |
| Temperature in the acceptance criterion is too low. | Log the acceptance rate; if it's near zero, the temperature is likely too low. | Increase the temperature parameter in the Monte Carlo acceptance step to allow more uphill moves. |
Issue 2: Unacceptable Computational Expense
| Symptoms | Possible Causes | Diagnostic Steps | Solutions |
|---|---|---|---|
| Single iteration takes very long. | The objective function is expensive to evaluate. | Use a profiler to confirm the objective function is the bottleneck. | Use a surrogate model (e.g., a Gaussian Process) to approximate the function. |
| The local minimization requires too many function evaluations. | Check the convergence report from the local minimizer. | Implement a budget for the maximum number of function calls per local minimization. | |
| Many iterations are required to converge. | Perturbations are too small. | Calculate the acceptance rate; a very high rate (>70%) may indicate small, ineffective steps. | Use a larger step size for perturbations to jump between basins more effectively. |
| Parallelize the evaluation of the population members, as they are often independent. |
Experimental Protocols & Methodologies
Protocol 1: Standard Basin-Hopping Workflow This protocol outlines the core steps of the standard, single-solution Basin-Hopping algorithm, which forms the foundation for BHPOP [2] [6].
- Initialization: Start with an initial random configuration, ( X_0 ).
- Local Minimization: Perform a local energy minimization from ( X0 ) to find the corresponding local minimum, ( \tilde{X0} ), with energy ( E(\tilde{X_0}) ). Set the current state to this minimum.
- Perturbation: Generate a new trial configuration, ( X{\text{new}} ), by applying a random perturbation to the current state ( \tilde{X}{\text{current}} ). This often involves random displacements to atomic coordinates or other degrees of freedom.
- Local Minimization: Perform a local minimization on ( X{\text{new}} ) to find its local minimum, ( \tilde{X}{\text{new}} ), and energy ( E(\tilde{X}_{\text{new}}) ).
- Acceptance/Rejection: Accept or reject the new minimum ( \tilde{X}_{\text{new}} ) based on the Metropolis criterion:
- If ( E(\tilde{X}{\text{new}}) \leq E(\tilde{X}{\text{current}}) ), accept the move.
- If ( E(\tilde{X}{\text{new}}) > E(\tilde{X}{\text{current}}) ), accept the move with probability ( \exp\left( -\frac{E(\tilde{X}{\text{new}}) - E(\tilde{X}{\text{current}})}{kB T} \right) ), where ( kB T ) is a temperature-like parameter.
- Update: If the move is accepted, set ( \tilde{X}{\text{current}} = \tilde{X}{\text{new}} ).
- Termination Check: Repeat steps 3-6 until a termination criterion is met (e.g., a maximum number of iterations or no improvement over a set number of steps).
Standard Basin-Hopping (BH) Algorithm Workflow
Protocol 2: Implementing a Population-Based BH (BHPOP) This protocol modifies the standard algorithm by introducing a population, enhancing its robustness [24].
- Initialization: Generate an initial population of ( N ) random configurations.
- Local Minimization: Perform local minimization on each member to create a population of local minima, ( P = {\tilde{X}1, \tilde{X}2, ..., \tilde{X}_N} ).
- Generation Loop: For each generation: a. Create Offspring: For each member in the population, create a new trial configuration by applying a perturbation. b. Local Minimization: Locally minimize each trial configuration. c. Selection: Form a new population by selecting the best ( N ) unique structures from the combined set of old population and new offspring. (Alternative selection strategies like tournament selection can also be used).
- Termination: Repeat Step 3 until a stopping condition is met.
Population-Based Basin-Hopping (BHPOP) Workflow
The Scientist's Toolkit: Research Reagent Solutions
| Item / Concept | Function / Role in the Experiment |
|---|---|
| Associative Memory Hamiltonian (AMH) | A coarse-grained molecular mechanics potential used as the energy function (( V(X )) for protein structure prediction. It incorporates bioinformatic data to guide the search towards physically realistic and evolutionarily favored structures [2]. |
| Metropolis Monte Carlo Criterion | The probabilistic rule used to decide whether to accept a new, higher-energy configuration. This allows the algorithm to escape local minima by occasionally taking "uphill" steps on the energy landscape, controlled by a temperature parameter (( k_B T )) [2] [6]. |
| Local Minimization Algorithm | A deterministic algorithm (e.g., L-BFGS, Conjugate Gradient) used to "quench" a perturbed configuration to the bottom of its current basin of attraction. This step transforms the original rugged energy landscape into a landscape of basin energies [2] [24]. |
| Objective Function (( f(x) )) | The "black box" function to be minimized. In the context of this thesis, it is typically a potential energy function for an atomic or molecular system. The algorithm only requires function values and, for efficient minimization, gradients [24]. |
| Significant Structures (SSBH) vs. Raw Structures (RSBH) | Two variants of the core algorithm. SSBH, where the current state is updated to the minimized geometry, is generally recommended over RSBH, where the state remains the pre-minimization geometry, for its superior performance [6]. |
Frequently Asked Questions
1. How do I implement constraints in a basin-hopping optimization?
You must pass the constraints correctly within the minimizer_kwargs dictionary. A common error is passing the variable names as strings instead of the variables themselves. The correct approach is shown below [64]:
cons object should be a dictionary or a list of dictionaries defining your constraints. For methods like L-BFGS-B that support bounds, you can pass them separately via minimizer_kwargs [12].
2. Why does my basin-hopping result violate constraints?
The basin-hopping algorithm can sometimes accept a new step based on the Metropolis criterion even if the local minimizer subsequently fails to find a constrained minimum from that starting point. To prevent this, implement a custom accept_test function. This function should check if a new solution satisfies your constraints before it is accepted [65].
3. Which local minimization methods support constraints in basin-hopping?
Not all local minimizers can handle constraints. When defining minimizer_kwargs, you must choose a method that supports constraints, such as SLSQP or trust-constr [65]. The default L-BFGS-B method only supports bounds.
4. What is the best way to enforce a minimum distance between optimized variables? You can define a nonlinear constraint. For instance, to ensure variables in a 3-parameter set are at least 0.1 apart in sorted order [65]:
This constraint function is designed to work with ordered coefficients and helps the minimizer by providing a single scalar value to satisfy.5. How do I choose the step size for perturbations?
The stepsize parameter controls the maximum random displacement applied to coordinates before each local minimization. It should be chosen based on your problem's characteristics. As a rule of thumb, it should be large enough to move the solution to a new "basin" of attraction but not so large that it behaves like a random search [12]. Some implementations allow for automatic adjustment of the step size to meet a target acceptance ratio [27].
Troubleshooting Guides
Problem: Basin-hopping returns a result that violates constraints.
| Step | Action | Code Example / Check |
|---|---|---|
| 1. Verify Local Minimizer | Ensure the local minimization method specified in minimizer_kwargs supports constraints. |
Use method='SLSQP' or 'trust-constr' instead of the default 'L-BFGS-B' [65]. |
| 2. Implement Acceptance Test | Add a custom accept_test to reject trial moves that violate constraints, regardless of their energy [65]. |
```python |
def accepttest(xnew, kwargs):
# Check all constraints
for con in constraints:
if con'fun' < 0.0: # For 'ineq' type
return False
return True
``|
| 3. Check Constraint Definition | Confirm your constraints are defined as a list of dictionaries and passed correctly. |constraints = [{'type': 'ineq', 'fun': lambda x: x[5] - x[4]}]` [64] |
Problem: The optimization is stuck in a local minimum and is not exploring globally.
| Step | Action | Code Example / Check |
|---|---|---|
| 1. Adjust Step Size | Increase the stepsize parameter to allow for larger perturbations, helping the algorithm "jump" to new basins [12]. |
result = basinhopping(..., stepsize=10.0) |
| 2. Enable Jumping | Use the "Basin Hopping with Occasional Jumping" algorithm. After a number of rejected steps, it performs larger jumps [27]. | Configure options like jump_max and jump_steps if supported by your implementation. |
| 3. Review Temperature | The "temperature" in the Monte Carlo acceptance criterion affects exploration. A higher temperature accepts more uphill moves early on. | This is often part of the algorithm's internal setup but can sometimes be adjusted via T parameter [2]. |
Experimental Protocols & Methodologies
Protocol 1: Implementing a Distance Constraint for Sorted Coefficients
This protocol is useful for enforcing a sequence in parameters, such as ensuring atomic radii or energy levels maintain a minimum separation [65].
- Define the Constraint Function:
- Formulate for Minimizer:
- Configure Basin-Hopping:
Protocol 2: Umbrella Sampling with Basin-Hopping for Landscape Exploration
This advanced protocol uses a biasing potential to explore regions of the energy landscape that might be hidden behind high barriers, commonly used in protein folding studies [2].
- Define an Order Parameter (Q): This parameter measures similarity to a target structure. For example, in proteins, it can be the fraction of native contacts.
- Apply a Biasing (Umbrella) Potential: This potential restrains the simulation to a specific range of the order parameter.
- Modify the Objective Function: The total energy minimized at each basin-hopping step becomes the sum of the original energy and the biasing potential.
- Run Serial Simulations: Perform multiple independent basin-hopping runs, each with a different center
Qifor the umbrella potential. The results can later be combined using weighted histogram analysis method (WHAM) to reconstruct the unbiased free energy landscape.
Table 1: Performance Comparison of Global Optimizers in a Surrogate Model
This table compares different global optimization algorithms applied to a surrogate model for a transcritical power cycle design, highlighting the trade-off between result quality and computational efficiency [66].
| Optimization Method | Levelized Cost of Energy (LCOE) [$/MWh] | Computational Time Reduction vs. Physics-Based Model |
|---|---|---|
| Physics-Based Model (Baseline) | 77.190 | Baseline |
| Surrogate-Dual Annealing (DA) | 77.912 | ~99% less |
| Surrogate-Basin Hopping (BH) | 78.876 | ~99% less |
Table 2: Basin-Hopping Algorithm Parameters and Their Effects
| Parameter | Default Value | Description | Impact on Optimization |
|---|---|---|---|
niter |
100 [12] | Number of basin-hopping iterations. | Higher values lead to more exhaustive search but longer compute times. |
stepsize |
0.5 [12] | Maximum displacement for random perturbation. | Too small: stuck in a local region. Too large: poor convergence. |
T |
1.0 (analogous) | "Temperature" for Metropolis acceptance criterion [2]. | Higher T promotes exploration; lower T promotes exploitation. |
target_ratio |
0.5 [27] | Target acceptance rate for step size adjustment. | Helps automatically tune stepsize for efficient exploration. |
Basin-Hopping with Constraints Workflow
The following diagram illustrates the workflow for implementing constraints in the basin-hopping algorithm, integrating checks at both the perturbation and local minimization stages.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Components for Basin-Hopping Experiments
| Item / Solution | Function / Role | Example / Notes |
|---|---|---|
| Coarse-Grained Potential | Reduces computational cost by grouping atoms; creates a smoother energy landscape. | Associative Memory Hamiltonian (AMH) [2], Gō-model [2]. |
| Local Minimizer (SLSQP) | A local optimization algorithm capable of handling nonlinear constraints. | Used inside minimizer_kwargs [65]. |
| Order Parameter (Q) | A collective variable that quantifies progress towards a target state. | Used in umbrella sampling to track similarity to native protein structure [2]. |
| Umbrella Potential | A biasing potential that restricts sampling to a specific range of an order parameter. | E(Q) = k * (Q - Q₀)⁴; allows exploration of specific reaction coordinates [2]. |
| Acceptance Test Function | A user-defined function to accept/reject a step based on custom criteria (e.g., constraints). | Critical for ensuring final solutions adhere to all physical or system constraints [65]. |
Techniques for Tracking Explored Regions and Ensuring Search Diversity
Frequently Asked Questions (FAQs)
FAQ 1: Why is tracking explored regions in a Basin-Hopping search important? Tracking explored regions is crucial because the Basin-Hopping algorithm is stochastic, meaning it does not guarantee finding the global minimum in a finite search. By keeping a record of explored local minima, researchers can direct the algorithm toward unexplored regions of the Potential Energy Surface (PES), avoid redundant calculations, and gain confidence that a diverse set of low-energy structures has been sampled. This helps in building a more representative set of structures for comparison with experimental data [4].
FAQ 2: What is a common cause of poor search diversity in Basin-Hopping?
A common cause is the algorithm becoming kinetically trapped in a local potential minimum. This often occurs when the simulation temperature (T) is set too low, preventing the acceptance of steps that temporarily increase energy, which is necessary to escape funnels leading to the current local minimum. In some cases, searches can be non-ergodic regardless of temperature, especially if the system requires changes in atomic connectivity that the default step-taking routine does not permit [4].
FAQ 3: How can I tell if my Basin-Hopping simulation is kinetically trapped?
You can monitor the callback function or the optimization result's history. If the global minimum candidate remains unchanged for a large number of iterations (which can be tracked using the niter_success parameter) and the algorithm consistently rediscover the same set of local minima, it is a strong indicator of being trapped [10] [4].
Troubleshooting Guides
Problem 1: Redundant Sampling and Inefficient Exploration
Symptoms
- The algorithm repeatedly finds the same local minima.
- The global minimum candidate does not improve over many iterations.
- A low number of unique minima are found relative to the total number of iterations (
niter).
Solution: Implement a Similarity Tracking and Clustering Protocol This solution uses techniques from unsupervised machine learning to identify and filter unique minima [4].
Experimental Protocol
- Define a Similarity Function: After each local minimization in the Basin-Hopping cycle, calculate a similarity metric between the new minimum and all previously found minima. A common function is the root-mean-square deviation (RMSD) of atomic coordinates after optimal alignment.
- Set a Uniqueness Threshold: Define a threshold value for the similarity metric (e.g., an RMSD of 0.5 Å). If the new minimum's similarity to any existing minimum is below this threshold, it is considered unique and added to the database.
- Employ Hierarchical Clustering: Periodically, use the database of found minima to perform hierarchical clustering. This creates a dendrogram that visually maps the relationships between all minima identified, revealing densely sampled regions and gaps in the PES exploration.
- Guide Future Steps: Use the clustering results to inform the
take_steproutine. If a new step leads to a region that is already densely sampled (as per the clusters), you can choose to reject the step prior to minimization or adjust thestepsizeto encourage a larger jump.
Resolution Verification The solution is working when the number of unique minima in the database increases steadily, and the dendrogram shows a diverse set of clusters rather than one dense cluster.
Problem 2: Kinetic Trapping in a Local Minimum
Symptoms
- The search consistently returns a low-energy structure that disagrees with experimental observations.
- The algorithm fails to find known minima from literature or other methods.
Solution: Adjust Algorithm Parameters and Step-Taking Routine
Solution A: Optimize Temperature and Stepsize
- Temperature (
T): The "temperature" parameter controls the acceptance of steps that increase energy. IfTis too low, the algorithm cannot escape local minima.Tshould be comparable to the typical function value difference between local minima [10]. Start with a higherTand reduce it if the search becomes too random. - Stepsize: This controls the maximum random displacement. The
stepsizeshould be comparable to the typical separation between local minima [10]. Basin-hopping can auto-adjust this, but providing a sensible initial value speeds up convergence. The adjustments are made everyintervaliterations based on thetarget_accept_rate[10].
Solution B: Customize the Acceptance Test
Use the accept_test parameter to define a custom test. For instance, you can create a test that forcefully accepts a step if the current minimum has not changed after a certain number of iterations, thus forcing an escape from a deep funnel [10].
Solution C: Implement a Directed Step-Taking Routine
For molecular systems, a custom take_step function can be more effective than random displacement. Instead of perturbing all Cartesian coordinates randomly, focus on specific degrees of freedom known to lead to conformational changes, such as dihedral angles in a molecule [4]. This can lead to more productive steps and prevent dissociations.
Table 1: Key Basin-Hopping Parameters for Managing Search Diversity
| Parameter | Description | Effect on Search Diversity | Recommended Tuning Range |
|---|---|---|---|
Temperature (T) |
Controls acceptance probability of higher-energy steps via Metropolis criterion [10]. | Low T: Risk of trapping. High T: Random, undirected search. |
Comparable to typical energy difference between local minima. |
| Stepsize | Maximum displacement for random perturbation [10]. | Small stepsize: Local exploration. Large stepsize: Global jumps, but may reduce efficiency. |
Comparable to typical separation between minima; auto-adjusted by algorithm. |
niter_success |
Stops run if global minimum candidate is unchanged for this number of iterations [10]. | Low value: May stop before finding true global minimum. High value: Allows more time to escape deep funnels. | Problem-dependent; use to break out of persistent traps. |
target_accept_rate |
The target for the algorithm's step acceptance rate, used to adjust stepsize [10]. |
A lower rate leads to larger stepsizes, promoting exploration. A higher rate does the opposite. | Default is 0.5; adjusting ±0.1 can influence exploration/exploitation balance. |
Table 2: Similarity Metrics for Tracking Explored Regions
| Metric | Description | Application Context | Uniqueness Threshold Example |
|---|---|---|---|
| Root-Mean-Square Deviation (RMSD) | Measures the average distance between atoms after optimal alignment [4]. | Comparing molecular clusters or protein conformations. | 0.5 - 1.0 Å for atomic clusters. |
| Q Value | Sequence-dependent measure of structural similarity for proteins, based on Cα pairwise contacts [2]. | Comparing protein folding states to a native structure. | 0.1 - 0.2 change in Q (range 0-1). |
| Custom Distance Function | A problem-specific function, d(A,B), that is non-negative, symmetric, and zero only for identical structures [4]. |
General purpose for any optimization problem where RMSD may not be suitable. | Defined by the specific function and system. |
Experimental Protocols
Protocol 1: Hierarchical Clustering of Sampled Minima This methodology is used to analyze the results of a Basin-Hopping search and identify unexplored regions [4].
- Data Collection: Run a Basin-Hopping simulation, storing the coordinates and energy of every accepted local minimum.
- Compute Similarity Matrix: Calculate a pairwise similarity matrix for all saved minima using a metric like RMSD.
- Perform Clustering: Use a hierarchical clustering algorithm (e.g., Ward's method) on the similarity matrix to generate a dendrogram.
- Analysis: The dendrogram reveals families of similar structures. Sparse branches indicate regions of the PES that may require further exploration. This information can guide the initiation of new, targeted Basin-Hopping runs.
Protocol 2: Interpolation for Transition State Searching This protocol helps identify the transition states connecting local minima, which is key to understanding kinetics [4].
- Select Endpoints: Choose two local minima (A and B) suspected to be connected.
- Interpolate Path: Generate a series of intermediate geometries along a path between A and B. This can be a linear interpolation in Cartesian coordinates or a more sophisticated path.
- Use as Guess for TS Search: Each intermediate geometry can be used as an initial guess for a transition state search algorithm (e.g., dimer method or Nudged Elastic Band).
- Validate: Confirm that the found transition state has exactly one imaginary frequency and connects to the two original minima.
Workflow Visualization
Research Reagent Solutions
Table 3: Essential Computational Tools for Basin-Hopping Research
| Item / Software | Function in Research |
|---|---|
SciPy (scipy.optimize.basinhopping) |
Provides the core Basin-Hopping algorithm implementation, handling the iteration loop, step acceptance, and local minimization calls [10]. |
| Local Minimizer (e.g., L-BFGS-B) | The algorithm used for the local minimization step within Basin-Hopping. Critical for efficiently finding the bottom of the local basin [10]. |
| Similarity / Clustering Library (e.g., SciPy) | Provides functions for calculating RMSD and performing hierarchical clustering, enabling the tracking of explored regions [4]. |
| Potential Energy Surface (PES) Model | The mathematical function describing the system's energy (e.g., molecular mechanics force field, associative memory Hamiltonian) [2]. This defines the landscape being searched. |
Custom take_step Function |
A user-defined routine to perturb coordinates in a problem-specific way, which can greatly improve search efficiency and diversity [10] [4]. |
Frequently Asked Questions
Q1: My Basin Hopping run seems to have stalled. How can I tell if it has truly converged or is just trapped? A true convergence is typically indicated by a persistent lack of improvement in the best-found energy over a large number of iterations, combined with a low acceptance rate for new trial steps. If the algorithm is trapped in a deep local minimum (not the global one), you might still see a low acceptance rate, but the energy value will be higher than the known global minimum for benchmark problems [12] [24]. Monitoring the trajectory of both the "current" and the "best" energy values is key. The "current" energy may fluctuate due to the Metropolis criterion accepting some uphill moves, but the "best" energy should show a staircase-like descent towards a stable value [9].
Q2: How many Basin Hopping iterations are sufficient to be confident the global minimum was found?
There is no universal number, as it depends on the complexity of your potential energy surface. A common practice is to run the algorithm for a fixed number of steps (e.g., 100 to 10,000, depending on the problem's computational cost) and perform multiple independent runs from different random starting points [12] [24]. If all or most runs converge to the same lowest energy structure, confidence is high. For very rugged landscapes, a higher number of iterations (niter) is necessary [12].
Q3: What is a good acceptance rate for new steps, and how does it relate to convergence? An acceptance rate that is too high (e.g., >80%) may indicate that the step size is too small, causing the search to be overly local and potentially missing other funnels. A rate that is too low (e.g., <20%) suggests the step size is too large, making it difficult to accept new minima and thus hindering progress. An adaptive Basin Hopping algorithm aims for a target acceptance rate of around 50% to balance exploration and refinement [9]. A dropping acceptance rate over time can be a sign of convergence.
Q4: How does Basin Hopping's performance compare to other global optimizers? Recent performance analyses show that Basin Hopping and its population-based variants are highly competitive. They are often almost as effective as state-of-the-art algorithms like Covariance Matrix Adaptation Evolution Strategy (CMA-ES) on standard benchmark functions and can be superior on difficult, real-world problems like atomic cluster energy minimization [24].
Troubleshooting Guides
Issue 1: Premature Convergence to a Local Minimum
Symptoms
- The best-found energy does not improve for a long time and is known to be higher than the global minimum.
- The acceptance rate for new steps becomes persistently very low.
Solutions
- Increase Perturbation Size: Adjust the
stepsizeparameter to be larger. This allows the algorithm to "jump" further away, potentially landing in the basin of attraction of a different, lower minimum [12]. A good rule of thumb is to set it to a meaningful fraction of your search space (e.g., 2.5%-5%) [12]. - Enable Adaptive Step Size: If your implementation supports it, use an adaptive step size control that dynamically adjusts the perturbation to maintain a healthy acceptance rate (e.g., near 50%) [9].
- Restart from Different Points: Execute multiple, independent Basin Hopping runs from randomly generated initial configurations. This is a simple and effective way to probe different regions of the energy landscape [24].
- Use a Population-Based Variant: Consider using a modified algorithm like BHPOP, which employs a population of walkers instead of a single one. This inherently provides a better exploration of the landscape [24].
Issue 2: Excessively Long Runtime
Symptoms
- The algorithm is taking too long to complete a single iteration or to find a satisfactory solution.
Solutions
- Tune the Local Minimizer: The local search (e.g., L-BFGS-B) is the most computationally expensive part. Ensure you are using an efficient local optimizer and have set its convergence tolerance (
tolorftol) to a sensible value—overly tight tolerances can waste resources [43]. - Implement Parallelization: A highly effective strategy is to perform multiple local minimizations in parallel at each Basin Hopping step. This "synched multi-search" approach explores the neighborhood more thoroughly per step and can significantly reduce wall-clock time on multi-core processors [9] [49].
- Reduce Problem Complexity: If possible, use knowledge of your system to reduce the number of degrees of freedom or use simplified, faster-to-compute models in the initial search phases.
Issue 3: Inconsistent Results Across Multiple Runs
Symptoms
- Different runs of the algorithm, with different random seeds, converge to different final energies and structures.
Solutions
- Increase the Number of Iterations: The inconsistency may stem from an insufficient sampling budget. Increase the
niterparameter to allow each run more time to escape local funnels and locate the global one [12]. - Check Step Size and Temperature: An overly large step size or a too-high temperature in the Metropolis criterion can lead to erratic behavior. Tuning these parameters or using an adaptive scheme can promote more consistent convergence [9].
- Report Statistical Results: For research purposes, it is good practice to perform a significant number of independent runs (e.g., 10-100) and report the success rate (the fraction of runs that found the global minimum) and the best, worst, and average performance [24].
Experimental Protocols & Data
Protocol 1: Standard Baseline Experiment
This protocol outlines a standard Basin Hopping run using common parameters from the literature [12].
- Initialization: Define the objective function (e.g., Lennard-Jones potential) and an initial atomic structure (e.g., a random cluster or a known configuration).
- Parameter Setup:
- Set the number of iterations (
niter) to 200. - Set the step size (
stepsize) for atomic displacements to 0.5 (in reduced units for the problem). - Set the temperature (
T) for the Metropolis criterion to 1.0. - Configure the local minimizer (e.g., L-BFGS-B from SciPy) with a force tolerance (
ftol) of 1e-5.
- Set the number of iterations (
- Execution: Run the Basin Hopping algorithm.
- Data Collection: Log the energy and coordinates at each step, noting which steps were accepted.
- Analysis: Plot the energy versus step number to visualize convergence.
Protocol 2: Performance Benchmarking vs. Other Algorithms
This protocol describes how to compare Basin Hopping against other optimizers fairly [24].
- Problem Selection: Choose a set of benchmark functions (e.g., from the BBOB suite) or a real-world problem like LJ38 cluster optimization.
- Algorithm Configuration: Configure Basin Hopping, Differential Evolution (DE), Particle Swarm Optimization (PSO), and CMA-ES using their recommended default parameters from reputable libraries.
- Budgeting: Define a fixed budget of function evaluations (e.g., 10,000) for all algorithms.
- Multiple Runs: Execute each algorithm 15 times on each benchmark function from different random initializations.
- Performance Measurement: Record the best-found function value for each run. Analyze and compare the results using performance profiles or data profiles, which show the fraction of problems solved versus the computational budget.
Table 1: Key Basin Hopping Parameters and Their Effects
| Parameter | Typical Value/Range | Function | Effect of Increasing the Parameter |
|---|---|---|---|
niter |
100 - 10,000 [12] | Total number of Monte Carlo steps. | Increases search duration and probability of success, but also computational cost. |
stepsize |
Problem-dependent (e.g., 0.5) [12] | Maximum displacement for random perturbation. | Promotes exploration of distant regions but may reduce the acceptance rate. |
T (Temperature) |
1.0 (common default) | Controls the probability of accepting uphill moves via the Metropolis criterion. | Allows more escape from local minima but can make convergence noisier. |
Local Minimizer ftol |
1e-3 to 1e-5 [43] | Force tolerance for local convergence. | Makes local minimization more precise but slower. Looser tolerances speed up the search. |
| Number of Parallel Candidates | 2 - 8 [9] [49] | Number of simultaneous local searches per step. | Greatly improves robustness and speed on multi-core CPUs, with near-linear speedup. |
Table 2: "Research Reagent Solutions" – Essential Computational Tools
| Item | Function/Benefit | Example Use-Case |
|---|---|---|
SciPy (basinhopping) |
A standard, accessible implementation of Basin Hopping in Python [12]. | Quick prototyping and testing of the algorithm on new academic problems. |
| Adaptive BH Code | Implements dynamic step size control to maintain a ~50% acceptance rate, improving efficiency [9]. | Tackling problems with unknown or complex landscape ruggedness. |
| L-BFGS-B Minimizer | A highly efficient local optimization algorithm for problems with bounds, commonly used within BH [12] [43]. | The local search workhorse for most BH runs on continuous potential energy surfaces. |
| ASE (Atomic Simulation Environment) | A Python package that provides various optimizers (BFGS, FIRE) and filters for structure optimization, including cell volume/shape [43]. | Performing structural relaxations in material science and computational chemistry. |
| Parallel BH Implementation | A version that runs multiple local minimizations concurrently, drastically reducing time-to-solution [9] [49]. | Running expensive searches on high-performance computing (HPC) clusters. |
Workflow Visualization
The following diagram illustrates the core workflow of the Basin Hopping algorithm and the key points for assessing convergence.
Key Diagnostic Plots for Convergence
To effectively assess convergence, monitor these visualizations during and after your Basin Hopping runs [9]:
- Energy Trajectory: Plot the energy of both the trial minima (blue lines) and the accepted minima (red points) at every step. A healthy search shows the accepted minima making a staircase descent, with trial energies fluctuating widely. The best-so-far energy (a green line) should plateau at the suspected global minimum.
- Acceptance Rate Over Time: Plot the moving average of the acceptance rate. A sudden, persistent drop may indicate convergence or trapping. An adaptive algorithm will show this rate oscillating around a target value (e.g., 50%).
- Lowest Energy Evolution: A plot showing only the best energy found as a function of the number of function evaluations. This is useful for comparing the performance and convergence speed of different algorithms or parameter sets [24].
Benchmarking Basin-Hopping: Performance Analysis Against Established Metaheuristics
This guide supports researchers in selecting and implementing global optimization algorithms for identifying local minima, a critical task in fields like computational chemistry and drug development. We focus on the Basin-Hopping (BH) method and compare it with three other prominent algorithms: Simulated Annealing (SA), Genetic Algorithms (GA), and the Covariance Matrix Adaptation Evolution Strategy (CMA-ES).
FAQ: Algorithm Selection and Comparison
1. What is the primary strength of the Basin-Hopping algorithm?
Basin-Hopping (BH) excels at navigating rugged potential energy surfaces (PES) by transforming the landscape into a collection of "basins of attraction." Its core strength lies in its two-step process: a random perturbation of the current structure followed by a local minimization. This "hopping" between local minima allows it to efficiently escape shallow traps and identify low-energy configurations, making it particularly effective for atomic cluster optimization and molecular structure prediction [9] [4] [2].
2. When should I choose CMA-ES over a Genetic Algorithm?
CMA-ES is often superior to GA on complex, non-separable, and ill-conditioned problems. While both are population-based, CMA-ES automatically learns the topology of the objective function by adapting an internal covariance matrix, similar to an inverse Hessian in quasi-Newton methods. This makes it highly efficient for correlated variables without requiring tedious parameter tuning. Empirical studies show CMA-ES can find optimal solutions faster and more reliably than GA-based tools in tasks like indoor daylight optimization [67] [68].
3. My BH search is trapped in a local minimum. How can I improve its exploration?
BH can suffer from kinetic trapping. Several strategies can enhance its exploration capabilities:
- Adaptive Step Size: Implement a step-size that dynamically adjusts based on the recent acceptance rate to balance broad exploration and local refinement [9].
- Parallel Evaluation: Generate and minimize multiple trial structures concurrently at each step. This reduces wall-clock time and decreases the probability of prolonged trapping [9].
- Machine Learning Augmentation: Integrate unsupervised learning techniques, such as similarity indices and hierarchical clustering, to track explored regions of the PES and guide the search toward unexplored areas [4].
4. How do these algorithms handle uncertainty and noisy objective functions?
CMA-ES and BH have inherent advantages in noisy or uncertain environments. CMA-ES is a derivative-free method that relies on the ranking of candidate solutions, making it robust to noise [68]. For BH, an "Uncertainty-Inclusive" variant (Un-BH) has been developed. This approach uses methods like Latin Hypercube Sampling to generate numerous random initial points for the deterministic optimization, helping to avoid convergence to false local optima caused by uncertainty [5].
5. What are the key computational expenses for each algorithm?
The primary computational cost varies by algorithm, as summarized in the table below.
| Algorithm | Primary Computational Cost | Key Cost Driver |
|---|---|---|
| Basin-Hopping (BH) | Local minimization steps [9] [2] | Number of energy/gradient evaluations per "hop" |
| Simulated Annealing (SA) | Function evaluations at sequential temperature steps [5] | Slow cooling schedule required for good results |
| Genetic Algorithm (GA) | Fitness evaluation of large populations over many generations [5] [67] | Population size and number of generations |
| CMA-ES | Function evaluations for population sampling [69] [68] | Population size and problem dimensionality |
Troubleshooting Guides
Issue 1: Poor Convergence or Stagnation in Basin-Hopping
Problem: The algorithm fails to find lower minima and appears stuck in a specific region of the energy landscape.
Possible Causes and Solutions:
- Cause: Inappropriate step size.
- Solution: Implement an adaptive step size control. Adjust the perturbation magnitude every 10-20 steps to maintain a target acceptance rate (e.g., 50%) [9].
- Cause: Insufficient exploration.
- Solution 1: Enable parallel evaluation. Use multiple CPU cores to minimize several candidate structures simultaneously at each BH step [9].
- Solution 2: Augment BH with machine learning. Use similarity functions and clustering to identify and avoid over-sampling similar minima, guiding the search to new regions [4].
- Cause: Low-level model chemistry is inaccurate.
- Solution: Use BH as a pre-screening tool with a fast, low-level potential (e.g., molecular mechanics), then refine the best structures with higher-level methods like density functional theory (DFT) [9].
Issue 2: Selecting an Algorithm for a New Problem
Problem: A researcher is unsure which optimization algorithm to use for their specific application.
Decision Workflow: The following diagram outlines a logical process for selecting the most suitable algorithm based on your problem's characteristics.
Issue 3: Algorithm-Specific Performance Tuning
Problem: An algorithm has been selected, but its performance and convergence are suboptimal.
Configuration Guidelines: Refer to the following protocol for tuning each algorithm's key parameters.
| Algorithm | Key Parameters | Recommended Tuning Protocol |
|---|---|---|
| Basin-Hopping | Step size, Temperature, Local minimizer | Start with an adaptive step size targeting a 50% acceptance rate. Set the Metropolis temperature (T) to 1.0 in reduced units. Use an efficient local minimizer like L-BFGS-B [9]. |
| CMA-ES | Population size (λ), Initial step size | Use the default parameters for most problems. For global search, employ a restart strategy with increasing population size (e.g., doubling λ after each restart). The initial step size should reflect the estimated distance to the optimum [68]. |
| Simulated Annealing | Cooling schedule, Initial temperature | The cooling schedule must be slow enough to allow for equilibrium. The initial temperature should be high enough to permit exploration of the search space [5]. |
| Genetic Algorithm | Population size, Mutation/ Crossover rates | Performance is highly dependent on these parameters. A larger population aids global search but increases computational cost. Mutation and crossover rates often require problem-specific experimentation [67]. |
The Scientist's Toolkit: Research Reagent Solutions
This table details key computational tools and methodologies essential for implementing the discussed optimization algorithms.
| Tool / Method | Function in Optimization | Example in Context |
|---|---|---|
| L-BFGS-B Minimizer | A quasi-Newton local optimization algorithm. | Used within each BH step for efficient local energy minimization after a perturbation [9]. |
| Lennard-Jones Potential | A simple model potential for pairwise atomic interactions. | Serves as a standard benchmark for evaluating BH and other algorithms on atomic clusters [9]. |
| Similarity Index & Clustering | Machine learning techniques to quantify geometric similarity. | Augments BH by identifying unique minima and mapping explored regions of the PES to avoid redundant searches [4]. |
| Latin Hypercube Sampling | A statistical method for generating a near-random sample of parameter values. | Used in Uncertainty-inclusive BH (Un-BH) to generate diverse initial points, mitigating the risk of poor convergence [5]. |
| Covariance Matrix | A core internal model in CMA-ES that captures variable dependencies. | Adapted iteratively to learn a second-order model of the objective function, guiding the search distribution [69] [68]. |
This technical support center provides essential resources for researchers employing the Basin-hopping global optimization algorithm. The Basin-hopping method is a powerful stochastic algorithm designed to find the global minimum of a function, particularly effective for rugged, multimodal energy landscapes common in fields like chemical physics, material science, and drug development [24] [12]. This guide addresses frequent experimental challenges, offers detailed protocols, and summarizes key performance metrics to support your research on local minima identification.
Key Performance Metrics in Basin-hopping
Tracking the right performance metrics is crucial for evaluating the effectiveness of your Basin-hopping experiments and comparing results with other optimization methods. The table below summarizes the core quantitative metrics used in performance analyses.
Table 1: Key Performance Metrics for Basin-hopping Experiments
| Metric Category | Specific Metric | Description and Role in Performance Analysis |
|---|---|---|
| Success Rate | Global Minimum Identification | The primary indicator of success is reliably finding the known global minimum, especially on benchmark functions [24]. |
| Success Rate on Real-World Problems | Performance on difficult, real-world problems (e.g., cluster energy minimization) tests robustness outside synthetic benchmarks [24]. | |
| Computational Effort | Number of Function Evaluations (NFEV) | A crucial budget metric; counts how many times the objective function is called, often used in fixed-budget analyses [24] [12]. |
Number of Iterations (niter) |
The total number of basin-hopping cycles, each comprising a perturbation and local minimization [70]. | |
| Computational Time | Wall-Clock Time | The total real time for a simulation; can be significantly reduced via parallel evaluation of candidate structures [9]. |
| Speedup from Parallelization | The reduction in wall-clock time achieved by running multiple local minimizations concurrently [9]. | |
| Algorithm Efficiency | Acceptance Rate | The rate at which new local minima are accepted. An adaptive stepsize can target a 50% rate to balance exploration and refinement [70] [9]. |
| Lowest Energy Found | Tracks the best solution identified over time, showing the algorithm's convergence progress [9]. |
Experimental Protocols and Methodologies
Standard Basin-hopping Algorithm Protocol
The following workflow details the core steps of the Basin-hopping algorithm, which combines global stepping with local minimization.
Figure 1: The Basin-Hopping Optimization Workflow. This diagram illustrates the iterative cycle of perturbation, local minimization, and acceptance that defines the algorithm.
- Initialization: Begin with an initial guess for the coordinates,
x0[70]. - Perturbation: In each iteration, apply a random perturbation to the current coordinates. The maximum amount of change is controlled by the
stepsizeparameter [70] [12]. - Local Minimization: Perform a local minimization from the perturbed coordinates. The default minimizer is often L-BFGS-B, but others like Nelder-Mead can be specified [70] [12].
- Acceptance Test: Accept or reject the new local minimum based primarily on the Metropolis criterion. The new minimum is always accepted if its function value is lower. If it is higher, it is accepted with a probability of
exp( -(f_new - f_old) / T ), whereTis a "temperature" parameter [70]. - Termination: The algorithm repeats steps 2-4 for a specified number of iterations (
niter) or until another convergence criterion is met [70].
Protocol for Performance Comparison Studies
To ensure fair and meaningful comparisons between Basin-hopping and other metaheuristics (e.g., Differential Evolution, CMA-ES), follow this established methodology [24]:
- Benchmark Selection: Use standardized test suites, such as the BBOB (Black-Box Optimization Benchmark) functions within environments like the IOHprofiler [24].
- Experimental Modes:
- Fixed-Budget Analysis: Run all algorithms for a predetermined number of function evaluations and compare the quality of the best solution found [24].
- Fixed-Target Analysis: Measure the number of function evaluations or computational time required by each algorithm to find a solution of a specific target quality [24].
- Real-World Validation: Supplement synthetic benchmarks with tests on real-world problems, such as molecular cluster energy minimization, to assess practical robustness [24].
- Statistical Reporting: Perform multiple independent runs for each algorithm and problem instance. Report aggregate results, such as medians and interquartile ranges, to account for stochastic variations [24].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software and Computational Tools for Basin-hopping Research
| Tool Name | Environment/Language | Primary Function and Application |
|---|---|---|
scipy.optimize.basinhopping |
Python/SciPy | A standard, readily available implementation for general numerical optimization problems [70] [12]. |
| Basinhopping.jl | Julia | A Julia implementation of the algorithm, offering flexibility and integration with the Julia technical computing ecosystem [26]. |
| Custom Adaptive BH | Python (NumPy, SciPy) | A customized implementation, like the one described by Carmona et al., featuring adaptive step size control and parallel local minimization for accelerated performance on complex landscapes like Lennard-Jones clusters [9]. |
| GMIN | Fortran | A free software program implementing Basin-hopping and many of its advanced variants [70]. |
| IOHprofiler | Framework | A benchmarking environment used for rigorous performance analysis and comparison of optimization algorithms on test suites like BBOB [24]. |
Troubleshooting Common Experimental Issues
FAQ 1: How do I choose thestepsizeandTparameters?
Choosing the stepsize and temperature T is critical for algorithm performance.
- Step Size (
stepsize): This parameter should be comparable to the typical separation between local minima in your search space. If set too small, the algorithm cannot escape the current basin. If too large, the search becomes random. The SciPy implementation can automatically adjust the stepsize to target an acceptance rate (e.g., 0.5), but providing a sensible initial guess speeds up convergence [70]. A good rule of thumb is to set it to a value that represents a meaningful perturbation in the context of your problem's domain [12]. - Temperature (
T): This parameter should be comparable to the typical function value difference between local minima. A higherTmakes the algorithm more likely to accept uphill moves, facilitating exploration. SettingT=0turns the algorithm into a greedy "Monotonic Basin-Hopping" where only downhill moves are accepted [70]. Trial runs on a simpler version of your problem can help calibrate this value.
FAQ 2: My optimization is stuck in a local minimum. What can I do?
- Increase Step Size: A larger
stepsizeallows for bigger jumps, potentially enabling the search to escape the current funnel of attraction and find a new basin [70]. - Increase Temperature: A higher temperature
Tincreases the probability of accepting solutions that are worse than the current best, helping the algorithm traverse energy barriers [70]. - Use a Population-Based Variant: Implement or switch to a population-based Basin-hopping method (BHPOP). This approach runs multiple Basin-hopping chains in parallel, improving the exploration of the search space and reducing the risk of all copies being trapped in the same local minimum [24].
- Customize the Perturbation: Implement a custom
take_stepfunction. Instead of a simple random displacement, you can design problem-specific moves that more effectively explore the configuration space. For example, in atomic systems, using delocalized internal coordinates can help preserve favorable bonding patterns [71] [70].
FAQ 3: How can I reduce the computational time of my Basin-hopping simulations?
- Parallel Local Minimization: A highly effective strategy is to generate several perturbed candidates in one iteration and perform their local minimizations concurrently on multiple CPU cores. The best minimum among them is then used in the acceptance test. This can lead to a nearly linear reduction in wall-clock time [9]. The following diagram illustrates this parallel workflow.
Figure 2: Parallel Basin-Hopping Workflow. This accelerated version generates and minimizes several candidates simultaneously, significantly reducing computation time.
- Use a Faster Local Minimizer: The choice of local minimizer can impact speed. While L-BFGS-B is a common default, testing other algorithms available in
scipy.optimize.minimizemight yield better performance for your specific objective function [12]. - Optimize Your Objective Function: Since the objective function is called thousands of times, ensuring it is computationally efficient is paramount. Consider code optimization, vectorization, or using just-in-time (JIT) compilers.
FAQ 4: How can I be more confident that I have found the true global minimum?
- Multiple Independent Runs: Execute the algorithm many times from different, random starting points. If all or most runs converge to the same lowest energy structure, confidence in that being the global minimum increases [70].
- Increase Iteration Count: Run the algorithm for a larger number of iterations (
niter). Theniter_successparameter can also be used to stop the run if the global minimum candidate remains unchanged for a specified number of iterations [70]. - Cross-Validation with Other Algorithms: Compare your results with those obtained from other well-established global optimization metaheuristics, such as Differential Evolution (DE) or Covariance Matrix Adaptation Evolution Strategy (CMA-ES) [24].
- Use Adaptive Methods: Implement an algorithm with adaptive step size control. This helps the search dynamically adjust its explorative behavior, preventing it from getting stuck and improving the thoroughness of the search over long runs [9].
Analysis on Standard Benchmark Functions (e.g., BBOB Test Suite)
Troubleshooting Guides and FAQs
Common Problem 1: Algorithm Trapped in Local Minima
Q: My basin-hopping algorithm consistently converges to local minima instead of finding the global optimum on BBOB functions. What adjustments can I make?
A: This is a common issue when the algorithm's parameters don't match the function's characteristics. Try these solutions:
Adjust Step Size: The stepsize parameter controls the random displacement magnitude. For BBOB's search domain of [-5,5]^D, start with stepsize=2.5 (approximately 5% of the domain size) [12] [10]. If stepsize is too small, the algorithm cannot escape local basins; if too large, it behaves like random search.
Optimize Temperature Parameter: The temperature
Tin the Metropolis criterion should be comparable to the typical function value difference between local minima [10]. Start with T=1.0 and adjust based on the specific BBOB function category:- For highly multimodal functions (BBOB groups 4-5): Use higher T values (2.0-5.0)
- For unimodal functions (BBOB groups 1-3): Use lower T values (0.5-1.0)
Implement Adaptive Step Sizes: Enable the automatic stepsize adjustment by setting
target_accept_rate=0.5andstepwise_factor=0.9[10], which allows the algorithm to dynamically optimize displacement sizes.Combine with Population Methods: Recent research shows that population variants of basin-hopping (BHPOP) perform better on difficult multimodal BBOB functions [24].
Common Problem 2: Poor Performance on Ill-Conditioned Functions
Q: Why does basin-hopping perform poorly on ill-conditioned BBOB functions like the Bent Cigar (f12) or Sharp Ridge (f13)?
A: Ill-conditioned functions have highly elongated basins that challenge coordinate-based perturbation:
Enhanced Local Minimizer: Configure
minimizer_kwargsto use better-suited local optimizers:The L-BFGS-B method handles ill-conditioning better than the default [10].
Coordinate System Rotation: Implement a custom
take_stepfunction that occasionally applies random rotations to the coordinate system, making it easier to navigate elongated valleys.Increased Iterations: For ill-conditioned BBOB functions (f10-f14), increase
niterto 500-1000 to allow sufficient exploration time [24].
Common Problem 3: Inconsistent Results Across BBOB Instances
Q: I get good results on some BBOB function instances but poor performance on others. How can I improve algorithm robustness?
A: BBOB functions have multiple instances with different optimal locations and values [72]:
Multiple Restarts: Implement an outer restart loop as shown in BBOB experimentation guidelines [73]:
Instance-Aware Parameter Tuning: Analyze performance across instances 1-15 (for tuning) separately from 16-110 (for validation) as recommended in BBOB best practices [72].
Custom Acceptance Tests: Implement an
accept_testfunction that incorporates domain knowledge about the BBOB function properties, such as rejecting solutions outside the theoretical global minimum region.
Common Problem 4: Excessive Computational Time
Q: Basin-hopping requires too many function evaluations to reach target accuracy on BBOB functions. How can I improve efficiency?
A: Optimization time is critical in benchmarking:
Local Minimizer Budget Control: Set strict tolerance limits in local minimization:
Dimensionality Scaling: Adjust parameters for high-dimensional BBOB problems (D=20,40). For dimension D, use
niter=100*Dandstepsize=5.0/sqrt(D)to maintain reasonable performance [24].Early Termination: Use
niter_successto stop if no improvement occurs after a set number of iterations, and implement the callback function to monitor progress.
Common Problem 5: Handling BBOB's Transformations and Symmetry Breaking
Q: How does basin-hopping cope with BBOB's asymmetric and non-linearly transformed functions?
A: BBOB applies transformations like Tasy (asymmetry) and Tosz (oscillation) that create irregular landscapes [72]:
Asymmetry Adaptation: For functions with T_asy transformation (creating asymmetric features), increase stepsize variability by setting
stepwise_factor=0.8to encourage diverse step directions.Oscillation Compensation: For T_osz transformed functions, implement a custom perturbation strategy that accounts for the oscillatory nature of the transformation in the region near the optimum.
Global Structure Exploitation: Despite transformations, most BBOB functions retain underlying global structure. Use moderate temperature values (T=1.0-2.0) to balance exploration of this structure with exploitation of local basins.
Experimental Protocols for Basin-Hopping on BBOB
Standard Benchmarking Protocol
Purpose: Fair comparison of basin-hopping variants against established algorithms on BBOB test suite.
Setup:
- Test Functions: All 24 noiseless BBOB functions [74] [72]
- Dimensions: D = 2, 3, 5, 10, 20, 40 [75]
- Instances: Use instances 1-15 for algorithm tuning, 16-110 for validation [73]
- Evaluation Budget: maxfuncevals = 100 * D² [73]
- Performance Measure: Expected Running Time (ERT) to reach target value ftarget = fopt + 10^(-8) [72]
Basin-Hopping Configuration:
Execution Steps:
- Initialize for each (function, dimension, instance) triple
- Run multiple independent trials with different random seeds
- Record best function value after each evaluation
- Compute ERT across all instances for each function group
- Compare with BBOB reference algorithms (CMA-ES, DE, PSO) [24]
Parameter Sensitivity Analysis Protocol
Purpose: Determine optimal parameter settings for basin-hopping across different BBOB function types.
Experimental Design:
- Test Subset: Representative functions from each BBOB group:
- f1 (Sphere) - separable
- f8 (Rosenbrock) - moderate conditioning
- f12 (Bent Cigar) - high conditioning
- f15 (Rastrigin) - multimodal with global structure
- f20 (Schwefel) - multimodal with weak global structure
Parameter Ranges:
- T: [0.1, 0.5, 1.0, 2.0, 5.0]
- stepsize: [0.1, 0.5, 1.0, 2.5, 5.0]
- niter: [50, 100, 200, 500]
- Local minimizers: ['L-BFGS-B', 'BFGS', 'Nelder-Mead']
Analysis Method:
- Latin Hypercube sampling for parameter combinations
- 10 independent runs per configuration
- Performance profiling to identify robust parameter settings
Table 1: BBOB Function Categories and Basin-Hopping Performance Characteristics
| BBOB Group | Function Numbers | Key Characteristics | Recommended BH niter | Recommended BH T | Expected ERT Range (D=10) |
|---|---|---|---|---|---|
| Separable | f1-f5 | Easy, convex or linear | 50-100 | 0.5-1.0 | 10^2-10^3 |
| Low/Moderate Conditioning | f6-f9 | Unimodal, moderate conditioning | 100-200 | 1.0-2.0 | 10^3-10^4 |
| High Conditioning Unimodal | f10-f14 | Ill-conditioned, unimodal | 200-500 | 1.0-2.0 | 10^4-10^5 |
| Multimodal Adequate Structure | f15-f19 | Many local minima, global structure | 300-500 | 2.0-5.0 | 10^4-10^5 |
| Multimodal Weak Structure | f20-f24 | Difficult, weak global structure | 500-1000 | 5.0+ | 10^5+ |
| Algorithm | Overall BBOB Rank | Best Performance On | Worst Performance On | Implementation Complexity |
|---|---|---|---|---|
| Basin-Hopping (Standard) | 3 | Unimodal functions (f1-f14) | Weak-structure multimodal (f20-f24) | Low |
| BHPOP (Population BH) | 2 | Mixed on all categories | - | Medium |
| CMA-ES | 1 | High conditioning functions | Separable functions | High |
| Differential Evolution | 4 | Multimodal functions | Unimodal functions | Medium |
| Particle Swarm Optimization | 5 | Early progress | Precision convergence | Medium |
Table 3: Optimal Basin-Hopping Parameters for BBOB Function Types
| Function Type | Stepsize | T | niter | Local Minimizer | Special Considerations |
|---|---|---|---|---|---|
| Separable | 1.0 | 0.5 | 50 | L-BFGS-B | Low iterations needed |
| Ill-conditioned | 2.5 | 1.0 | 200 | L-BFGS-B | Strict tolerance important |
| Multimodal with structure | 3.0 | 3.0 | 300 | Nelder-Mead | Higher T helps escape |
| Multimodal weak structure | 4.0 | 5.0 | 500 | BFGS | Population variants recommended |
Workflow Visualization
Basin-Hopping BBOB Evaluation Workflow
Algorithm Performance Comparison Diagram
Research Reagent Solutions
Table 4: Essential Computational Tools for Basin-Hopping BBOB Research
| Tool/Resource | Purpose | Usage in Research | Source |
|---|---|---|---|
| COCO Platform | Automated benchmarking | Running BBOB experiments, performance tracking | [76] [77] |
| SciPy basinhopping | Core algorithm implementation | Baseline BH algorithm, customization | [10] |
| BBOB Function Definitions | Standard test problems | Algorithm validation, comparison | [74] [72] |
| IOH Profiler | Experimental data collection | Performance profiling, data logging | [24] |
| DEAP BBOB Interface | Evolutionary algorithm framework | Comparison algorithms, BBOB integration | [73] |
| Optuna-BBOB Wrapper | Hyperparameter optimization | Parameter tuning for BH | [75] |
Table 5: Algorithm Variants and Extensions for Enhanced Performance
| Variant | Key Features | Best For BBOB Groups | Implementation Complexity |
|---|---|---|---|
| Standard Basin-Hopping | Basic BH with coordinate perturbation | 1-3 (unimodal) | Low [12] [10] |
| BHPOP | Population-based BH | 4-5 (multimodal) | Medium [24] |
| Adaptive Step BH | Automatic stepsize adjustment | All groups, especially 4-5 | Medium [10] |
| Monotonic BH (T=0) | Only accepting improving steps | 1-2 (easy unimodal) | Low [10] |
| Custom Step BH | Problem-specific perturbations | Specific function types | High |
For researchers in drug development and computational chemistry, navigating the complex energy landscapes of molecular systems to find the most stable structures is a fundamental challenge. The basin-hopping algorithm is a powerful global optimization technique designed specifically for this task. This guide provides a detailed overview of its strengths, weaknesses, and optimal application scenarios to inform your experimental design.
FAQ: Core Concepts and Applications
What is the basin-hopping algorithm? Basin-hopping is a global optimization technique that transforms a potential energy surface into a collection of basins. It iterates through a cycle of random perturbation of coordinates, local minimization, and acceptance or rejection of the new coordinates based on the minimized function value using a Metropolis criterion [10] [25]. It is designed to mimic the natural process of energy minimization of clusters of atoms [10].
For what types of problems is basin-hopping best suited? Basin-hopping excels in problems with "funnel-like, but rugged" energy landscapes [10]. It has proven particularly effective for:
- Atomic and Molecular Clusters: Finding minimum energy structures of Lennard-Jones clusters [9] and boron or metal clusters [78].
- Drug Discovery: Exploring the configuration space of molecules, such as determining putative binding modes for cyclodextrin inclusion complexes [51].
- General Global Optimization: Solving difficult black-box optimization problems in high-dimensional spaces [24].
How does basin-hopping help in escaping local minima? The algorithm's core strength is its ability to escape local minima. It accepts not only steps that lower the energy but also, with a certain probability, steps that increase the energy. This probability is governed by a "temperature" parameter (T) and the Metropolis criterion. Accepting these "uphill" moves allows the search to escape the basin of attraction of a local minimum and explore new regions of the potential energy surface [10].
What are the key parameters in a basin-hopping simulation? The performance of basin-hopping is highly dependent on a few critical parameters [10]:
- Step Size: The maximum displacement for random perturbations. It should be comparable to the typical separation between local minima.
- Temperature (T): Controls the probability of accepting uphill moves. It should be comparable to the typical energy difference between local minima.
- Number of Iterations (
niter): The total number of basin-hopping steps.
Modern implementations often include an adaptive step size that is dynamically adjusted to maintain a target acceptance rate (e.g., 50%), which significantly improves usability and efficiency [9] [10].
Experimental Protocols and Workflows
Protocol: Standard Basin-Hopping Run for a Molecular System
This protocol outlines the steps for setting up and running a basin-hopping calculation to identify low-energy molecular conformations, as applied in studies like the analysis of cyclodextrin inclusion complexes [51].
Initial Structure Preparation:
- Obtain an initial molecular geometry, typically from a file format containing Cartesian coordinates (e.g., .molden, .xyz) [9].
- For drug discovery applications, this could be a ligand or a host-guest complex.
Algorithm Configuration:
- Local Minimizer: Select a local minimization algorithm. The L-BFGS-B method is a common choice for its efficiency [9]. Configure it within the
minimizer_kwargsparameter. - Step Size: Set an initial step size for perturbations. If using an adaptive method, define the
target_accept_rate(e.g., 0.5) andstepwise_factor(e.g., 0.9) [10]. - Temperature: Set the temperature
Tfor the Metropolis criterion. A value of 1.0 (in reduced units) is sometimes used as a starting point [9], but it should be chosen based on the system's energy scale. - Iterations: Define the number of basin-hopping iterations (
niter). Several hundred may be needed for complex systems [9].
- Local Minimizer: Select a local minimization algorithm. The L-BFGS-B method is a common choice for its efficiency [9]. Configure it within the
Execution and Monitoring:
- Run the calculation. The algorithm will cycle through perturbation, minimization, and acceptance.
- Monitor the acceptance rate and the evolution of the lowest energy found to assess progress [9].
Result Analysis:
- The output is a list of accepted low-energy structures and their energies.
- Analyze the structures to identify the global minimum and key low-lying local minima relevant to your research, such as stable conformations or binding modes [51].
The following diagram illustrates the control flow of this protocol.
Performance Comparison: Basin-Hopping vs. Other Metaheuristics
A 2024 performance analysis compared basin-hopping to other established metaheuristics on benchmark functions and real-world problems [24]. The key findings are summarized below.
Table 1: Performance Comparison of Basin-Hopping Against Established Metaheuristics (Adapted from [24])
| Algorithm | Acronym | Key Principle | Reported Performance vs. BH |
|---|---|---|---|
| Basin Hopping | BH | Perturbation + Local Minimization + Metropolis | Baseline |
| Covariance Matrix Adaptation Evolution Strategy | CMA-ES | Population-based, adaptive update of candidate distribution | Almost as good as BH on benchmarks, worse on real-world problems |
| Differential Evolution | DE | Population-based, vector differences for mutation | Not as good as BH |
| Particle Swarm Optimization | PSO | Population-based, social behavior of birds/fish | Not as good as BH |
| Population-Based BH | BHPOP | A population-based variant of BH | Comparable to CMA-ES on benchmarks, better on real-world problems |
Troubleshooting Common Experimental Issues
Problem: The calculation is converging to a local minimum, not the global minimum.
- Cause 1: Step size is too small. A small step size prevents the algorithm from escaping the current funnel of attraction.
- Cause 2: Temperature is too low. A low temperature makes the acceptance criterion too strict, rejecting all uphill moves.
- Solution: Increase the temperature
Tto be comparable to the function value difference between local minima in your system [10].
- Solution: Increase the temperature
- Cause 3: Insufficient number of iterations.
- Solution: Increase
niter. For complex systems, run the algorithm multiple times from different random starting points to ensure consistency [10].
- Solution: Increase
Problem: The calculation is running too slowly.
- Cause 1: The local minimizer is inefficient or converging poorly.
- Solution: In the
minimizer_kwargs, try a different minimization method (e.g., "BFGS") or adjust its tolerance settings [10].
- Solution: In the
- Cause 2: The system is large, and each energy evaluation is expensive.
- Solution: Implement parallelization. As demonstrated in recent work, multiple trial structures can be minimized concurrently, achieving nearly linear speedup for up to eight concurrent processes [9].
Problem: The results are not reproducible.
- Cause: The algorithm uses random numbers for perturbations.
- Solution: Set the random number generator seed (
seedorrngparameter) to ensure the same sequence of random numbers is used in each run, leading to reproducible results [10].
- Solution: Set the random number generator seed (
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for Basin-Hopping Research
| Tool / Resource | Function / Purpose | Implementation Example |
|---|---|---|
| Local Optimizer | Finds the nearest local minimum from a given starting configuration. | L-BFGS-B, BFGS, conjugate gradient [9] [78]. |
| Potential Energy Surface (PES) Calculator | Computes the energy and forces for a given atomic configuration. | Lennard-Jones potential [9], density functional theory (DFT), machine-learned potentials [9] [78]. |
| Parallelization Framework | Accelerates the search by evaluating multiple candidates simultaneously. | Python's multiprocessing.Pool [9]. |
| Structure Visualization | Analyzes and interprets the resulting low-energy geometries. | Molden format output [9], AMSMovie GUI [47]. |
| Specialized Software | Provides pre-built, optimized implementations of the algorithm. | SciPy (scipy.optimize.basinhopping) [10], AMS PESExploration [47]. |
Decision Guide: When to Choose Basin-Hopping
Choosing the right optimization algorithm depends on the nature of your problem. The following flowchart outlines key decision points.
Key Strengths and Weaknesses
Strengths:
- Effective on Rugged Landscapes: Its two-phase nature makes it exceptionally good at handling complex, multimodal potential energy surfaces, a common challenge in molecular structure search [9] [24].
- Conceptual Simplicity: The algorithm is straightforward to understand and implement, making it a good choice for practitioners [24].
- Proven Reliability: It has a long track record of success in physics and chemistry, reliably finding global minima for challenging problems like Lennard-Jones clusters [9] [25].
- Parallelizability: Its efficiency can be significantly improved by evaluating multiple trial structures in parallel [9].
Weaknesses:
- Parameter Sensitivity: Performance can be sensitive to the choice of step size and temperature, though adaptive methods mitigate this [10].
- Dependence on Local Optimizer: Its efficiency is tied to the speed and reliability of the chosen local minimization algorithm [10].
- Risk of Overspecialization: Highly customized versions of basin-hopping for specific chemical systems may lose their general applicability [24].
- Not Always the Fastest: For some problem types, particularly those where population-based methods excel, other algorithms like CMA-ES might be competitive or superior [24].
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: My Basin-Hopping search seems trapped in a high-energy region and cannot find structures that match my experimental spectrum. What should I do?
A1: This is a common issue with non-ergodic sampling. Implement these strategies:
- Adaptive Step Size: Use an algorithm that dynamically adjusts the perturbation step size based on the acceptance rate to better balance exploration and refinement. A target acceptance rate of 50% is often effective [79] [10].
- Parallel Candidate Evaluation: Generate and minimize multiple trial structures concurrently at each step. This reduces wall-clock time and improves the probability of escaping deep local minima [79].
- Machine Learning Guidance: Employ unsupervised machine learning techniques, such as similarity indices and hierarchical clustering, on the collected local minima. This helps identify under-sampled regions of the potential energy surface (PES) and directs the search towards new, unexplored areas [4] [80].
Q2: How can I ensure the low-energy structures found by Basin-Hopping are actually relevant to my experimental ion mobility or spectroscopy data? A2: The global minimum is not always the most experimentally relevant structure. To address this:
- Identify Kinetically Accessible Minima: Locate not just the local minima, but also the transition states that connect them. This allows you to model the thermodynamic accessibility of different isomers/conformers under experimental conditions [4] [80].
- Compare to a Spectrum of Minima: Match your experimental data (e.g., collision cross-section, IR spectrum) against a collection of low-lying local minima, not just the single global minimum. The experimental ensemble often includes several thermally accessible structures [4].
Q3: My computational collision cross-sections (CCS) consistently differ from my experimental TWIMS values. Is my search method flawed? A3: The search method might not be the primary issue. Focus on the validation pipeline:
- Calibration is Critical: CCS values can vary significantly (>2% difference) across different IMS platforms (e.g., DTIMS vs. TWIMS). Ensure you are using a calibration standard and protocol appropriate for your specific instrument [81].
- Understand Method Limitations: CCS calculations from structures are often based on molecular mechanics, which may not be accurate enough for certain systems. If resources allow, refine low-energy candidates from the BH search with higher-level quantum mechanical calculations [4].
Q4: How do I choose the optimal "temperature" (T) and "stepsize" parameters for my Basin-Hopping run? A4: These parameters are system-dependent.
- Temperature (T): The parameter
Tin the Metropolis criterion should be comparable to the typical energy difference between local minima on your PES. IfTis too low, the search may get trapped; if too high, it becomes a random search. For best results,Tshould be comparable to the separation (in function value) between local minima [10]. - Step Size: The initial step size should be comparable to the typical spatial separation between local minima. Modern implementations often include an adaptive scheme that automatically adjusts the step size to maintain a target acceptance rate (e.g., 0.5) [79] [10].
Troubleshooting Common Experimental Validation Issues
The table below summarizes common problems, their potential causes, and solutions when validating Basin-Hopping results with spectroscopic and ion mobility data.
| Problem | Potential Cause | Solution |
|---|---|---|
| No structural match to experimental IR spectrum | Inaccurate force field; Non-ergodic sampling missing key isomers; Incorrect protonation state. | Refine candidates with higher-level (e.g., DFT) calculations; Use augmented BH with multiple searches; Manually check key isomers (e.g., prototropic forms) [4] [80]. |
| Poor correlation between computed and experimental CCS values | Incorrect instrument calibration; Inadequate structural sampling; Limitations in CCS calculation model. | Use correct calibration standard for instrument (e.g., Agilent Tune Mix); Increase BH iterations/parallel candidates; Use a validated CCS calculation software (e.g., MOBCAL, IMOS) [81]. |
| Algorithm fails to find the known global minimum | Poor parameter choice (T, stepsize); Stochastic nature of the algorithm. | Use adaptive step size; Run multiple BH searches from different random starting points; Increase number of iterations (niter) [79] [10]. |
| High computational cost for large systems | Serial evaluation of candidates; Expensive energy/force calculations. | Use parallel evaluation of candidate structures; Start with machine-learned potentials or force fields before switching to ab initio methods [79]. |
Experimental Protocols for Method Validation
Protocol 1: Validating Structures via Infrared Multiple Photon Dissociation (IRMPD) Spectroscopy
This protocol outlines the steps for assigning an IRMPD spectrum using structures generated from a Basin-Hopping search, as applied in the case study of the protonated serine dimer [4] [80].
1. Conduct a Comprehensive BH Search:
- System Setup: Define the molecular system, ensuring correct protonation states and isomeric forms.
- BH Execution: Perform an augmented BH search using an appropriate level of theory (e.g., molecular mechanics). Key parameters may include a temperature for the Metropolis criterion and an adaptive step size.
- Data Collection: Save all unique local minima identified during the search, not just the lowest-energy structure.
2. Post-Processing and Clustering:
- Calculate Similarity: Use a geometric similarity function (e.g., root-mean-square deviation) to quantify differences between all saved minima [4] [80].
- Cluster Structures: Apply hierarchical clustering to group nearly identical structures, providing a non-redundant set of unique conformers/isomers for further analysis [4] [80].
3. Spectral Prediction and Matching:
- Quantum Chemical Refinement: Select the lowest-energy structure from each major cluster and refine its geometry using density functional theory (DFT).
- Theoretical Spectrum Calculation: Compute the harmonic vibrational frequencies and IR intensities for each refined structure.
- Assignment: Compare the theoretical IR spectra of the low-energy candidates with the experimental IRMPD spectrum to identify the spectral carriers.
Protocol 2: Validating Structures via Ion Mobility Collision Cross-Section (CCS)
This protocol describes how to determine the temperature-dependent CCS of a molecule, such as protonated alanine tripeptide, using BH results [4] [81] [80].
1. Generate a Conformational Ensemble with BH:
- Perform a BH search, focusing on the degrees of freedom relevant to conformation (e.g., dihedral angles). The Metropolis temperature can be linked to the experimental temperature of interest [4].
2. Calculate Theoretical CCS Values:
- For the ensemble of unique low-energy structures, calculate the theoretical CCS for the relevant drift gas (e.g., N₂).
- Use established methods like the trajectory method in software such as MOBCAL or IMoS.
3. Compare with Experimental IM-MS Data:
- Instrument Calibration: Calibrate the IM-MS instrument using a known standard (e.g., Agilent Tune Mix) to ensure accurate drift time to CCS conversion [81].
- Data Acquisition: Measure the drift time and calculate the experimental CCS for the analyte under defined conditions (gas, temperature, electric field).
- Dynamic CCS Modeling: To model a temperature-dependent CCS, create a weighted average of the theoretical CCS values, where the weights are determined by the Boltzmann population of each conformer at the experimental temperature based on their relative energies [4].
The Scientist's Toolkit: Research Reagent Solutions
Table: Key Computational and Analytical Tools for BH Validation
| Item | Function / Description | Example Use Case |
|---|---|---|
| Basin-Hopping Algorithm | Global optimization method that transforms the PES into a set of interpenetrating staircases, combining random perturbation, local minimization, and Metropolis acceptance [4] [25]. | Core engine for finding low-energy minima of atomic clusters (e.g., Lennard-Jones) and molecular systems [79] [4]. |
| Similarity Function (d(A,B)) | A metric to quantify the geometric difference between two conformations (A and B) in the search space. Enables clustering and identification of unique minima [4] [80]. | Used to avoid redundant optimization and to guide the search towards unexplored regions of the PES [4]. |
| Local Minimizer (L-BFGS-B) | An efficient algorithm for local optimization of smooth functions with bounds on variables, often called at each step of the BH routine [79] [10]. | Performs the local minimization after each random perturbation in the BH cycle to find the bottom of the current energy basin [79]. |
| CCS Calibration Standard | A solution of known compounds with established CCS values used to calibrate the drift time-to-CCS conversion on an IM-MS instrument [81]. | Essential for obtaining accurate CCS values on platforms like the Agilent 6560 or MOBILion MOBIE for comparison with computed values [81]. |
| Hierarchical Clustering | An unsupervised machine learning technique used to group molecular structures based on their geometric similarity [4] [80]. | Post-processing BH results to obtain a non-redundant set of unique conformers for spectroscopic comparison [4]. |
Workflow Visualization
Reliability and Robustness Assessment for Black-Box Optimization Problems
Frequently Asked Questions (FAQs)
Q1: What does the "Serious error occurred during Optimization results processing" message mean, and how should I resolve it? This error indicates that an internal exception was triggered during the optimization process. The recommended action is to first check the relevant log files for detailed error information. If the cause is not apparent, you should contact the software's support team and provide them with the log files for further diagnosis [82].
Q2: My basin-hopping run is not converging to a consistent solution. How can I improve its reliability?
Non-convergence can often be mitigated by adjusting the algorithm's parameters. A key parameter is niter_success, which stops the run if the global minimum candidate remains unchanged for a specified number of iterations. Setting this parameter can prevent endless loops in rugged energy landscapes. Furthermore, ensuring that the stepsize is comparable to the typical separation between local minima and that the temperature T is comparable to the typical function value difference between minima can significantly enhance convergence behavior [29].
Q3: How can I trust the results from a black-box optimizer, especially in a high-stakes field like drug design? Building trust involves using frameworks designed for explainability and reliability. For instance, the IEMSO (Inclusive Explainability Metrics for Surrogate Optimization) framework provides model-agnostic metrics to enhance transparency. These metrics explain the sampling core, batch properties, the optimization process, and feature importance, allowing you to understand the rationale behind the optimizer's decisions before and after expensive evaluations [83]. Furthermore, robust frameworks like RABVI (Robust and Automated Black-box Variational Inference) incorporate automated techniques to detect inaccurate estimates and provide a termination criterion that balances accuracy and computational cost [84].
Q4: What is a practical way to handle analytical constraints in a black-box optimization problem? Some modern solvers allow you to incorporate analytical constraints directly into the model, just as you would in a classical optimization problem. This is more efficient than treating all constraints as black-box functions or using penalty methods. For example, in a revenue management problem with multiple periods, constraints ensuring that available units decrease over time can be added directly to guide the solver and improve feasibility [85].
Troubleshooting Guides
Common Basin-Hopping and Black-Box Optimization Issues
Table 1: Common Error Messages and Solutions
| Symptom / Error Message | Possible Cause | Troubleshooting Steps |
|---|---|---|
| "Unknown Product: |
Input data is missing, incorrectly referenced, or has improperly formatted XML tags [82]. | Verify the input file for correct spelling, capitalization, and spaces. Ensure all required XML tags are present and properly structured [82]. |
| "Check product weights and dimensions" | Zero values in the product’s length, width, height, or weight fields [82]. | Ensure all dimensional and weight fields contain valid non-zero values [82]. |
| "Exception caught from optimizer" | An internal exception was triggered by the optimization process [82]. | Check the detailed log files. If the error persists, contact technical support with the log files [82]. |
| Algorithm is trapped in a local minimum | The step size may be too small, or the acceptance criterion too strict [29]. | Implement a custom accept_test function to forcefully escape the local minimum. Adjust the stepsize and temperature T to encourage broader exploration [29]. |
| Poor performance in noisy optimization scenarios | Traditional rank-based optimizers are sensitive to noise [86]. | Use algorithms designed for noise robustness, such as those derived from natural policy gradients (e.g., an improved MORE algorithm), which optimize expected fitness directly without relying on rankings [86]. |
Quantitative Performance of Different Black-Box Optimizers
When dealing with computationally expensive black-box functions, the number of function evaluations is a critical resource. The following table summarizes the performance of different optimizers on a revenue management problem, showing the mean absolute gap from the best-known solution [85].
Table 2: Comparison of Black-Box Optimizers by Number of Function Evaluations [85]
| Method | 25 Evaluations | 50 Evaluations | 75 Evaluations | 100 Evaluations | 125 Evaluations | 150 Evaluations |
|---|---|---|---|---|---|---|
| Random Search | 56.59 | 41.68 | 32.93 | 28.22 | 21.44 | 21.44 |
| RBFOpt | 169.91 | 36.89 | 11.05 | 7.56 | 5.55 | 2.31 |
| NOMAD | 27.34 | 21.00 | 12.32 | 1.53 | 0.33 | 0.06 |
| Hexaly 10.0 | 10.29 | 0.06 | 0 | 0 | 0 | 0 |
Experimental Protocols for Reliability Assessment
Protocol: Evaluating Optimization Reliability using RABVI Framework
The RABVI (Robust and Automated Black-box VI) framework provides a method to improve the reliability of stochastic optimization [84].
- Adaptive Learning Rate: Implement a mechanism that detects the convergence of fixed-learning-rate iterates and adaptively decreases the learning rate.
- Divergence Estimation: Estimate the symmetrized Kullback-Leibler (KL) divergence between the current variational approximation and the optimal one. This serves as a quantitative measure of convergence quality.
- Termination Criterion: Employ a novel termination criterion that compares (i) the predicted relative decrease in the symmetrized KL divergence if a smaller learning rate were used against (ii) the predicted computation required to converge with the smaller rate. This allows users to balance desired accuracy against computational cost [84].
Protocol: Explainability and Trust Assessment using IEMSO Metrics
Apply the Inclusive Explainability Metrics for Surrogate Optimization (IEMSO) to gain transparency into the black-box optimization process [83].
- Categorize Metrics: Apply the four primary categories of IEMSO metrics:
- Sampling Core Metrics: Explain the reasoning behind individual sample point selection.
- Batch Properties Metrics: Detail the criteria and efficiency of generated sample batches, such as diversity.
- Optimization Process Metrics: Provide an overview of the entire process, including convergence.
- Feature Importance Metrics: Describe the importance of input features on the objective and the surrogate model's behavior.
- Gain Intermediate and Post-hoc Explanations: Use these metrics to interpret the optimizer's strategy both during the run (intermediate) and after it has completed (post-hoc) to build trust in the results [83].
Workflow and Relationship Visualizations
Basin-Hopping Optimization Workflow
Reliability Assessment Framework
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Key Computational Tools for Basin-Hopping and Black-Box Optimization
| Tool / Reagent | Function / Application | Context in Research |
|---|---|---|
SciPy basinhopping |
A Python implementation of the basin-hopping algorithm for global optimization [29]. | The primary algorithm for identifying low-energy molecular configurations and reaction intermediates in computational chemistry studies [25] [29]. |
| Surrogate Model (e.g., Gaussian Process) | A fast, approximate model of a costly black-box function, used for strategic sampling [83] [85]. | Crucial for optimizing expensive-to-evaluate functions, such as molecular binding affinity in drug design or complex simulation outputs [83] [85]. |
| IEMSO Framework | A set of model-agnostic metrics to provide explainability and build trust in surrogate optimization results [83]. | Used to interpret and validate the decisions of a black-box optimizer, ensuring that the proposed solutions are reliable and actionable for researchers [83]. |
| 2-HP-β-Cyclodextrin | A solubility enhancer used to form inclusion complexes with lipophilic drug candidates [51]. | A key reagent in case studies that optimize drug properties. Its use can alter ligand-receptor binding, a factor that must be considered when optimizing for in vitro activity [51]. |
| Molecular Fragmentation & Graph Theory | A computational method to reduce the dimensionality of potential energy surface sampling [87]. | Enhances the efficiency of basin-hopping for large molecular systems by breaking them into smaller fragments and analyzing results with graph theory [87]. |
Basin-Hopping (BH) has established itself as a powerful and versatile stochastic algorithm for global optimization, particularly renowned for its effectiveness in solving complex, multimodal problems in fields like chemical physics and drug development. The standard algorithm operates through an iterative two-phase process: a random perturbation of the current coordinates, followed by local minimization of the perturbed solution [88] [29]. The acceptance of the new minimum is traditionally governed by the Metropolis criterion, which allows the algorithm to escape local minima by sometimes accepting solutions with higher energy [12].
However, practitioners often encounter limitations with the standard BH algorithm, including the risk of becoming kinetically trapped in local minima and inconsistent performance across different energy landscapes [80] [89]. To overcome these challenges, researchers have developed innovative hybrid approaches that integrate the core BH framework with other algorithmic strategies. These hybrids leverage the strengths of each component, creating more robust and efficient tools for identifying global minima in computationally demanding research, such as molecular structure prediction and drug design [24] [1] [80]. This guide addresses the specific issues users may face when implementing these advanced techniques.
Troubleshooting Common Hybrid BH Implementation Issues
This section provides solutions to frequently encountered problems when integrating Basin-Hopping with other algorithms.
FAQ 1: My hybrid BH algorithm is not exploring new regions of the search space and seems trapped. How can I encourage more global exploration?
- Problem: The algorithm is exhibiting limited exploration, likely due to the standard random walk perturbation being too local to escape a large, deep basin or to jump between distant minima.
- Solution: Consider replacing the default perturbation step with a non-local "skipping" mechanism.
- Basin Hopping with Skipping (BH-S): This hybrid replaces the random walk step with a "skipping" perturbation [89]. Instead of a single random displacement, it generates a sequence of states in a straight line until it finds a point with lower energy than the current state or reaches a limit. This acts like a long-range probe, enabling direct hops between distant basins that would be difficult to reach via local moves [89].
- Augmentation with Unsupervised Learning: Incorporate techniques like similarity indices and hierarchical clustering to guide the search [80]. By quantifying the similarity between found local minima, you can identify and prioritize the exploration of under-sampled regions of the potential energy surface (PES), ensuring the algorithm does not waste cycles re-exploring known territory.
FAQ 2: My computational budget is limited, and the hybrid BH is too slow. How can I accelerate the search without sacrificing reliability?
- Problem: The local minimization step within BH is often the most computationally expensive part of the algorithm, creating a performance bottleneck.
- Solution: Implement a parallelized hybrid search strategy to perform multiple local minimizations simultaneously.
- Synched Multi-L-BFGS BH: A highly effective hybrid approach involves generating several random neighbors from the current point and launching independent local minimizers (e.g., L-BFGS) from each of them [49]. The best of the resulting minima is then used for the next BH step. This "deeper" exploration of the neighborhood significantly improves convergence speed and reliability on hard problems. The key advantage is that these local searches are embarrassingly parallel and can be distributed across multiple CPU cores or GPUs, leading to a substantial reduction in wall-clock time [49].
FAQ 3: How do I choose the right local minimizer and parameters for my specific problem when setting up a hybrid BH?
- Problem: The performance of a BH hybrid is highly sensitive to the choice of the local minimizer and its associated parameters.
- Solution: Select the local minimizer based on problem properties like dimensionality and the availability of gradient information.
- Standard Choices: The SciPy
basinhoppingfunction defaults to the "L-BFGS-B" method, which is a good general-purpose choice for problems with bounds [12] [29]. For problems without gradients, "Nelder-Mead" is a common alternative [12]. - Advanced Hybrids: For population-based BH variants (BHPOP), the performance has been shown to be competitive with state-of-the-art algorithms like Covariance Matrix Adaptation Evolution Strategy (CMA-ES) on synthetic benchmarks and can even outperform them on difficult real-world problems like cluster energy minimization [24]. Parameter tuning is crucial; the
stepsizeshould be comparable to the typical separation between local minima, and the temperatureTshould be comparable to the typical function value difference between them [88] [29].
- Standard Choices: The SciPy
FAQ 4: How can I make my BH search more efficient when dealing with a large number of similar local minima?
- Problem: The algorithm is redundantly optimizing and revisiting geometrically similar structures, wasting computational resources.
- Solution: Integrate a machine learning-based filtering step to identify and eliminate redundant minima.
- Similarity and Clustering: After each local minimization, compute a similarity metric (e.g., based on root-mean-square deviation of atomic coordinates for molecular problems) between the new minimum and all previously found minima [80].
- Workflow Integration: This similarity data can be used in real-time to reject duplicates before expensive post-processing (like frequency calculations) or to guide the perturbation step away from already densely sampled basins, making the overall PES mapping much more efficient [80].
Experimental Protocols for Key Hybrid BH Approaches
This section provides detailed, actionable methodologies for implementing the hybrid BH techniques discussed in the troubleshooting guide.
Protocol 1: Implementing Basin-Hopping with Skipping (BH-S)
Objective: To enhance the global exploration capability of the standard BH algorithm by implementing a non-local "skipping" perturbation [89].
- Initialization: Begin from a current state ( Xn ) and define the target set ( Cn ) as the sublevel set ( {x \in D: f(x) \le f(X_{n})} ) [89].
- Direction Generation: Sample a random direction ( \varphi ) from the unit sphere ( \mathbb{S}^{d-1} ).
- Radial Step Sampling: Sample a step size ( r ) from a probability distribution ( q_r(\cdot | \varphi) ) (e.g., a Gaussian or uniform distribution).
- Skipping Loop: Construct the candidate point ( Zj = Xn + j \cdot r \cdot \varphi ) for ( j = 1, 2, ... ).
- If ( Zj \in Cn ) (i.e., ( f(Zj) \le f(Xn) )), accept ( Yn = Zj ) and exit the loop.
- If the maximum number of skips is reached without entering ( Cn ), set ( Yn = X_n ).
- Local Minimization: Apply a local minimization routine (e.g., L-BFGS-B) to ( Yn ) to find a local minimum ( Un ) [89].
- Acceptance: Accept the new minimum ( U_n ) based on the Metropolis criterion or use a monotonic acceptance rule (always accept only lower values) [89].
The following diagram illustrates the core skipping workflow compared to a standard perturbation:
Protocol 2: Parallel Multi-Start BH with Synched Local Searches
Objective: To significantly reduce computation time and improve convergence reliability by parallelizing the local minimization phase of the BH algorithm [49].
- Initialization: Start from a current state ( X_n ).
- Parallel Perturbation: Generate ( K ) independent random perturbations around ( Xn ), creating a set of candidate points ( {Y{n,1}, Y{n,2}, ..., Y{n,K}} ).
- Parallel Local Minimization: Distribute the ( K ) candidate points to available CPU or GPU cores. On each core, run an identical local minimization algorithm (e.g., L-BFGS-B) on its assigned candidate point, producing a set of local minima ( {U{n,1}, U{n,2}, ..., U_{n,K}} ) [49].
- Synchronization and Selection: After all local minimizations complete, gather the results and select the best local minimum ( U{n,best} ) from the set ( {U{n,1}, ..., U_{n,K}} ).
- Acceptance: Use ( U_{n,best} ) in the standard BH acceptance step (Metropolis or monotonic) as the proposal for the next state [49].
Table 1: Performance Comparison of Standard BH vs. Parallel BH on Selected Benchmark Problems
| Test Function | Dimension | Success Rate (Standard BH) | Success Rate (Parallel BH, K=4) | Avg. Speedup |
|---|---|---|---|---|
| Levy | 10D | 65% | 95% | 2.8x |
| Ackley | 15D | 40% | 85% | 3.1x |
| Schwefel | 20D | 15% | 70% | 3.5x |
Note: Data is indicative and based on results from benchmarks of hard tests [49]. Actual performance is dependent on hardware and problem specifics.
Protocol 3: Augmenting BH with Machine Learning for Efficient PES Mapping
Objective: To reduce redundant computation and achieve broader coverage of the Potential Energy Surface (PES) by using ML to identify unique minima and guide exploration [80].
- Database Setup: Maintain a growing database ( \mathcal{D} ) of all unique local minima found during the BH search. Each entry stores the coordinates and the corresponding energy.
- Similarity Assessment: After each local minimization finds a new minimum ( U{new} ), calculate a similarity metric ( d(U{new}, Mi) ) between ( U{new} ) and every existing minimum ( M_i ) in ( \mathcal{D} ). For molecular systems, this could be the Root-Mean-Square Deviation (RMSD) of atomic coordinates [80].
- Uniqueness Check & Clustering: If ( d(U{new}, Mi) ) is below a defined threshold for any ( Mi ), classify ( U{new} ) as a duplicate and discard it. Periodically, use hierarchical clustering on ( \mathcal{D} ) to group minima into structurally similar families [80].
- Guided Perturbation: Use the clustering information to bias future perturbations. If the algorithm is trapped in a densely sampled basin, the perturbation step size can be increased, or a direction can be chosen that points towards a sparsely sampled region of the PES.
The Scientist's Toolkit: Essential Research Reagents & Solutions
This table details key computational tools and concepts essential for implementing hybrid BH approaches effectively.
Table 2: Key Research Reagents and Solutions for Hybrid BH Experiments
| Item / Concept | Function / Role in the Experiment | Example Implementation / Notes |
|---|---|---|
| Local Minimizer (L-BFGS-B) | The core engine for the local minimization phase. Efficiently finds the nearest local minimum using gradient information and handles bound constraints. | The default in SciPy's basinhopping. Crucial for performance [12] [29]. |
| Similarity Metric (e.g., RMSD) | A quantitative measure to compare two molecular structures or solution vectors, used to avoid redundant optimizations. | Essential for the ML-augmented BH protocol. RMSD is standard for molecular geometries [80]. |
| Unsupervised Clustering Algorithm | Groups found local minima into families based on structural similarity, providing insight into the PES landscape and guiding exploration. | Hierarchical clustering is commonly used for this purpose [80]. |
| Skipping Perturbation | A non-local move that enables the algorithm to jump between distant basins, improving global exploration capabilities. | Core component of the BH-S algorithm [89]. |
| Parallel Computing Framework | A software environment to execute multiple local minimizations simultaneously, drastically reducing total computation time. | Can be implemented using Python's multiprocessing or GPU-accelerated libraries [49]. |
Conclusion
The Basin-Hopping algorithm stands as a remarkably effective and accessible strategy for global optimization, particularly suited for the complex, high-dimensional potential energy surfaces encountered in drug discovery and biomolecular modeling. Its core strength lies in simplifying the search landscape by focusing on local minima, a approach that is further enhanced by modern adaptations like parallel processing, adaptive step-size control, and machine learning integration. When benchmarked against other metaheuristics, BH demonstrates competitive, and often superior, performance on difficult real-world problems like cluster energy minimization. For biomedical research, the future of BH is tightly coupled with its integration into multi-scale workflows, combining the speed of empirical potentials with the accuracy of ab initio methods, and its application in AI-driven drug discovery platforms for rapid lead identification and optimization. Embracing these advanced BH methodologies will be pivotal in reducing computational costs, improving predictive accuracy, and ultimately accelerating the development of novel therapeutics.
References
- 1. A Review of Global Optimization Methods for Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12537999/]
- 2. Protein structure prediction using basin-hopping - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2674628/]
- 3. basinhopping — SciPy v1.14.0 Manual [https://docs.scipy.org/doc/scipy-1.14.0/reference/generated/scipy.optimize.basinhopping.html]
- 4. Augmenting Basin-Hopping With Techniques From ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2019.00519/full]
- 5. Dimensionality reduction and uncertainty quantification for ... [https://www.sciencedirect.com/science/article/abs/pii/S0360544225031585]
- 6. An investigation of two approaches to basin hopping ... [https://www.sciencedirect.com/science/article/abs/pii/S000926149800431X]
- 7. Global optimization - Sahinidis - Georgia Institute of Technology [https://sahinidis.coe.gatech.edu/research/global-optimization]
- 8. Application of Optimization Algorithms in Chemistry [https://www.frontiersin.org/research-topics/9088/application-of-optimization-algorithms-in-chemistry/magazine]
- 9. Revisiting and Accelerating the Basin Hopping Algorithm ... [https://arxiv.org/html/2510.25938v1]
- 10. basinhopping — SciPy v1.16.2 Manual [https://docs.scipy.org/doc/scipy/reference/generated/scipy.optimize.basinhopping.html]
- 11. Scipy BasinHopping not returning correct global minima [https://stackoverflow.com/questions/73010732/scipy-basinhopping-not-returning-correct-global-minima]
- 12. Basin Hopping Optimization in Python [https://www.machinelearningmastery.com/basin-hopping-optimization-in-python/]
- 13. Discretized boundary-oriented progressive learning ... [https://www.sciencedirect.com/science/article/abs/pii/S0167278924003014]
- 14. Potential energy surface [https://en.wikipedia.org/wiki/Potential_energy_surface]
- 15. Potential Energy Surface [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Quantum_Mechanics/11%3A_Molecules/Potential_Energy_Surface]
- 16. CAMEO Chemicals Software [https://www.epa.gov/cameo/cameo-chemicals-software]
- 17. MARPLOT | response.restoration.noaa.gov [https://response.restoration.noaa.gov/oil-and-chemical-spills/chemical-spills/marplot]
- 18. How to make scipy.optimize.basinhopping find the global ... [https://datascience.stackexchange.com/questions/49255/how-to-make-scipy-optimize-basinhopping-find-the-global-optimal-point]
- 19. Constraints seem to be ignored using basinhopping with ... [https://stackoverflow.com/questions/52359147/constraints-seem-to-be-ignored-using-basinhopping-with-cobyla-method]
- 20. Example to understand scipy basin hopping optimization ... [https://stackoverflow.com/questions/22207372/example-to-understand-scipy-basin-hopping-optimization-function]
- 21. New bounds and formulations for the deterministic global optimization... [https://link.springer.com/article/10.1007/s10898-025-01476-7]
- 22. Trilateration-Based Multilevel Method for Minimizing the... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7302545/]
- 23. Lennard-Jones potential [https://en.wikipedia.org/wiki/Lennard-Jones_potential]
- 24. A Performance Analysis of Basin Hopping Compared to ... [https://arxiv.org/html/2403.05877v1]
- 25. Basin-hopping [https://en.wikipedia.org/wiki/Basin-hopping]
- 26. Basinhopping.jl [https://juliapackages.com/p/basinhopping]
- 27. Basin Hopping — EON: Long Timescale Dynamics [https://theory.cm.utexas.edu/eon/basin_hopping.html]
- 28. Programing and optimizing code for a Basin Hopping method [https://stackoverflow.com/questions/65875730/programing-and-optimizing-code-for-a-basin-hopping-method]
- 29. basinhopping — SciPy v1.16.2 Manual [https://docs.scipy.org/doc//scipy-1.16.2/reference/generated/scipy.optimize.basinhopping.html]
- 30. why scipy.optimize.basinhopping give different results [https://stackoverflow.com/questions/70703026/why-scipy-optimize-basinhopping-give-different-results]
- 31. Understanding the output of scipy.optimize.basinhopping [https://stackoverflow.com/questions/27728483/understanding-the-output-of-scipy-optimize-basinhopping]
- 32. Meaning of 'minimization_failures' when using scipy. ... [https://stackoverflow.com/questions/27411636/meaning-of-minimization-failures-when-using-scipy-optimize-basinhopping]
- 33. What is the Basin-Hopping Metropolis Temperature? [https://stackoverflow.com/questions/62784096/what-is-the-basin-hopping-metropolis-temperature]
- 34. Scipy_Basinhopping — Sherpa 4.18.0 documentation [https://sherpa.readthedocs.io/en/4.18.0/optimisers/api/sherpa.optmethods.optscipy.Scipy_Basinhopping.html]
- 35. Does basinhopping of scipy changes stepsize randomly or ... [https://stackoverflow.com/questions/30558116/does-basinhopping-of-scipy-changes-stepsize-randomly-or-adaptively-by-default]
- 36. Basin hopping optimization in Python [https://aicorespot.io/basin-hopping-optimization-in-python/]
- 37. Numerical optimization based on the L-BFGS method [https://towardsdatascience.com/numerical-optimization-based-on-the-l-bfgs-method-f6582135b0ca/]
- 38. An intro the BFGS Optimisation Algorithm [https://aicorespot.io/an-intro-the-bfgs-optimisation-algorithm/]
- 39. How to Use Nelder-Mead Optimization in Python [https://www.machinelearningmastery.com/how-to-use-nelder-mead-optimization-in-python/]
- 40. error using L-BFGS-B in scipy - python [https://stackoverflow.com/questions/18985048/error-using-l-bfgs-b-in-scipy]
- 41. glitch in numerical optimization in python using nelder-mead [https://stackoverflow.com/questions/12676745/glitch-in-numerical-optimization-in-python-using-nelder-mead]
- 42. BUG: optimize: worse behaviour of new L-BFGS-B ... [https://github.com/scipy/scipy/issues/23161]
- 43. Structure optimization — ASE documentation [https://wiki.fysik.dtu.dk/ase/ase/optimize.html]
- 44. Structural optimization of atomic clusters using iterated ... [https://www.sciencedirect.com/science/article/pii/S0010465525001572]
- 45. Optimization of SiO 2 with GHA and basin hopping [https://www.sciencedirect.com/science/article/abs/pii/S0927025621006984]
- 46. python 3.x - Scipy Optimize Basin Hopping fails [https://stackoverflow.com/questions/57853306/scipy-optimize-basin-hopping-fails]
- 47. Automated PES Exploration — AMS 2025.1 documentation [https://www.scm.com/doc/AMS/Tasks/PES_Exploration.html]
- 48. Revisiting and Accelerating the Basin Hopping Algorithm ... [https://arxiv.org/abs/2510.25938]
- 49. Basin Hopping with synched multi L-BFGS local searches. ... [https://www.sciencedirect.com/science/article/abs/pii/S0096300319301444]
- 50. How to parallelize scipy.optimize basinhopping? [https://stackoverflow.com/questions/79292593/how-to-parallelize-scipy-optimize-basinhopping]
- 51. Impact of 2-hydroxypropyl-β-cyclodextrin inclusion complex ... [https://www.sciencedirect.com/science/article/pii/S000629522400323X]
- 52. Exploration–exploitation dilemma [https://en.wikipedia.org/wiki/Exploration%E2%80%93exploitation_dilemma]
- 53. Exploitation and Exploration in Machine Learning [https://www.geeksforgeeks.org/machine-learning/exploitation-and-exploration-in-machine-learning/]
- 54. An improved FOX optimization algorithm using adaptive ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0331965]
- 55. Adaptive Step-Size Strategies [https://www.emergentmind.com/topics/adaptive-step-size-strategy]
- 56. A New Global Optimization Method for Complex Systems [https://pubmed.ncbi.nlm.nih.gov/15267429/]
- 57. Systematic Comparison of Genetic Algorithm and Basin ... [https://eprints.whiterose.ac.uk/id/eprint/186995/]
- 58. [cond-mat/9803344] Global Optimization by Basin-Hopping ... [https://arxiv.org/abs/cond-mat/9803344]
- 59. AIを活用し化学反応の理解を自動化する仕組みを開発 ―深層 ... [https://www.ims.ac.jp/news/2025/03/0314.html]
- 60. [BoTorch]Monte Carlo獲得関数とその最適化 [https://qiita.com/narrowlyapplicable/items/3c2c80e05e16fa935cf1]
- 61. ハイパーパラメーター最適化フレームワークOptunaの実装解説 [https://cyberagent.ai/blog/research/optuna-from-scratch]
- 62. 【PySCF】構造最適化・振動数計算のやり方【Pythonで ... [https://labo-code.com/python/quantum-chemical-calculation/pyscf-opt-freq/]
- 63. Quantum basin hopping with gradient-based local ... [https://ui.adsabs.harvard.edu/abs/2005quant.ph..7193B/abstract]
- 64. python - Declare constraints for basinhopping optimization [https://stackoverflow.com/questions/31278026/declare-constraints-for-basinhopping-optimization]
- 65. Best way to enforce a distance between coefficients [https://stackoverflow.com/questions/76507527/best-way-to-enforce-a-distance-between-coefficients]
- 66. Development of surrogate-optimization models for a novel ... [https://www.sciencedirect.com/science/article/pii/S2666546823000836]
- 67. Based Tool and Covariance Matrix Adaptation [https://experts.illinois.edu/en/publications/comparison-between-popular-genetic-algorithm-ga-based-tool-and-co/]
- 68. CMA-ES [https://cma-es.github.io/]
- 69. CMA-ES [https://en.wikipedia.org/wiki/CMA-ES]
- 70. basinhopping — SciPy v1.15.0 Manual [https://docs.scipy.org/doc/scipy-1.15.0/reference/generated/scipy.optimize.basinhopping.html]
- 71. Systematic Comparison of Genetic Algorithm and Basin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9126620/]
- 72. The bbob Test Suite [https://coco-platform.org/testsuites/bbob/overview.html]
- 73. Benchmarking Against the Bests (BBOB) - DEAP documentation [https://deap.readthedocs.io/en/master/tutorials/advanced/benchmarking.html]
- 74. bbob - COCO Home [http://numbbo.github.io/coco/testsuites/bbob]
- 75. The blackbox optimization benchmarking (bbob) test suite [https://hub.optuna.org/benchmarks/bbob/]
- 76. Workshops Current (BBOB-2025) [https://coco-platform.org/workshops/bbob2025.html]
- 77. BBOB Workshop Series - COCO Home [https://numbbo.github.io/workshops/]
- 78. Structure Search with the Strategic Escape Algorithm - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11983718/]
- 79. Revisiting and Accelerating the Basin Hopping Algorithm for Lennard–Jones Clusters: Adaptive and Parallel Implementation in Python [https://arxiv.org/html/2510.25938]
- 80. Augmenting Basin-Hopping With Techniques From ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6693329/]
- 81. Evaluating Ion Mobility Data Acquisition, Calibration, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12221786/]
- 82. Blackbox Optimization Error Messages [https://support.magiclogic.com/hc/en-us/articles/26147991250189-Blackbox-Optimization-Error-Messages]
- 83. Building Trust in Black-box Optimization: A Comprehensive ... [https://arxiv.org/html/2410.14573v1]
- 84. A Framework for Improving the Reliability of Black-box ... [https://arxiv.org/abs/2203.15945]
- 85. Solving Constrained Black-Box Optimization Problems [https://www.hexaly.com/tutorials/solving-constrained-black-box-optimization-problems]
- 86. Robust Black-Box Optimization for Stochastic Search and ... [https://jmlr.org/papers/v25/22-0564.html]
- 87. Efficient Basin - Hopping Sampling of Reaction Intermediates through... [https://pubmed.ncbi.nlm.nih.gov/26580762/]
- 88. scipy.optimize.basinhopping — SciPy v1.11.2 Manual [https://docs.scipy.org/doc//scipy-1.11.2/reference/generated/scipy.optimize.basinhopping.html]
- 89. Hopping between distant basins | Journal of Global ... [https://link.springer.com/article/10.1007/s10898-022-01153-z]