Benchmarking in Computational Chemistry: A Guide to Validating Models for Drug Discovery and Biomedical Research
This article provides a comprehensive guide to benchmarking in computational chemistry, a critical process for validating the accuracy and reliability of models that predict molecular properties and behaviors.
Benchmarking in Computational Chemistry: A Guide to Validating Models for Drug Discovery and Biomedical Research
Abstract
This article provides a comprehensive guide to benchmarking in computational chemistry, a critical process for validating the accuracy and reliability of models that predict molecular properties and behaviors. Tailored for researchers, scientists, and drug development professionals, it covers foundational concepts, methodological applications, troubleshooting strategies, and comparative validation techniques. By exploring current frameworks, statistical metrics, and real-world case studies—from predicting toxicokinetic properties to assessing machine-learned interatomic potentials—this resource offers practical insights for implementing robust benchmarking practices. The goal is to empower scientists to select and develop trustworthy computational tools that accelerate innovation in biomedical and clinical research.
What is Model Benchmarking? Core Concepts and Critical Importance in Computational Chemistry
In computational chemistry, benchmarking is the systematic process of evaluating and comparing the performance of computational models against reliable experimental data to assess their accuracy and reliability. This process serves as a critical bridge between theoretical predictions and real-world observations, establishing confidence in computational methods used for predicting molecular properties and behaviors. The fundamental purpose of benchmarking is to rigorously quantify how well a computational model reproduces physically observable phenomena, thereby guiding method selection, improvement, and establishing domains of applicability [1] [2].
Benchmarking differs from, yet complements, the broader concepts of verification and validation (V&V). Verification addresses whether a computational model is solved correctly ("solving the equations right"), while validation determines whether the correct model is being solved ("solving the right equations") [3] [2]. Benchmarking operates primarily within the validation domain, providing the empirical evidence needed to assess a model's physical accuracy. As computational simulations increasingly inform critical decisions in drug discovery and materials design, rigorous benchmarking has become indispensable for transitioning from qualitative demonstrations to quantitatively reliable predictions [4].
The Critical Importance of Benchmarking
Benchmarking provides the essential foundation for establishing credibility in computational models, particularly as these models are increasingly used to reduce reliance on costly physical experiments [3]. In high-consequence fields like drug development and nuclear safety, where computational predictions may inform regulatory decisions or safety assessments, comprehensive benchmarking is not merely academic but a practical necessity [2]. The benchmarking process creates a structured framework for method selection, enabling researchers to choose the most appropriate computational approach for their specific problem from among multiple competing methods [5].
The recent emergence of machine-learned interatomic potentials (MLIPs) highlights benchmarking's role in driving methodological progress. As noted in evaluations of models trained on the Open Molecules 2025 (OMol25) dataset, "trust is especially critical here because scientists need to rely on these models to produce physically sound results that translate to and can be used for scientific research" [6]. Benchmarking creates a competitive yet collaborative environment where "better benchmarks and evaluations have been essential for progress and advancing many fields of ML" [6]. This friendly competition, often facilitated by public leaderboards, accelerates innovation while maintaining rigorous standards [7] [6].
Furthermore, benchmarking identifies limitations and weaknesses in current methodologies, directing future development efforts. For instance, benchmarking revealed that MLIPs with similar training errors can exhibit significantly different performance on real-world tasks like molecular dynamics simulations [7]. Similarly, in drug discovery, benchmarking has exposed concerning inconsistencies in binding pose prediction, with one study finding that "only 26% of noncovalently bound ligands and 46% of covalent inhibitors could be accurately regenerated within 2.0 Å RMSD of the experimental pose" [4]. These performance gaps, uncovered through systematic benchmarking, highlight where methodological improvements are most urgently needed.
Key Components of a Benchmarking Framework
Defining Purpose and Scope
The first step in any benchmarking study involves clearly defining its purpose and scope. Studies generally fall into three categories: method development benchmarks (conducted by method developers to demonstrate advantages of a new approach), neutral benchmarks (independent comparisons of existing methods), and community challenges (organized competitions like CASP for protein structure prediction) [5]. The scope must balance comprehensiveness with practical constraints, ensuring the benchmark addresses chemically relevant questions without becoming unmanageably large [5].
Selection of Methods
Method selection should be guided by the benchmark's purpose. Neutral benchmarks should strive to include all available methods for a specific analysis, functioning as a comprehensive review, while method development benchmarks may compare against a representative subset of state-of-the-art and baseline methods [5]. Inclusion criteria should be clearly defined and applied consistently, such as requiring freely available software implementations that can be successfully installed without excessive troubleshooting [5].
Selection of Benchmark Datasets
The choice of reference datasets fundamentally influences benchmarking outcomes. Two primary dataset types are used: experimental data and simulated data. Experimental data provides real-world relevance but may have measurement uncertainties, while simulated data offers known "ground truth" but must accurately reflect real systems [5]. High-quality benchmarks employ diverse datasets representing various conditions and system types to thoroughly test method robustness [7]. For example, the MLIPAudit framework includes "organic small molecules, flexible peptides, folded protein domains, molecular liquids and solvated systems" to comprehensively evaluate machine-learned interatomic potentials [7].
Performance Metrics and Evaluation Criteria
Quantitative performance metrics enable objective method comparison. Common metrics in computational chemistry include Mean Absolute Error (MAE), Root Mean Square Error (RMSE), and the coefficient of determination (R²) [8] [1]. Different metrics emphasize different aspects of performance—MAE weights all errors equally, RMSE penalizes larger errors more heavily, and R² measures correlation strength. Selection should align with the benchmark's goals, often requiring multiple metrics to fully characterize performance [5].
Experimental Protocols and Methodologies
Benchmarking Neural Network Potentials
A recent benchmark evaluating OMol25-trained neural network potentials (NNPs) on experimental reduction potential and electron affinity data exemplifies rigorous methodology [8]. For reduction potential prediction, researchers obtained experimental data from a compiled dataset of 193 main-group and 120 organometallic species. The protocol involved:
- Geometry Optimization: Optimizing non-reduced and reduced structures for each species using each NNP with geomeTRIC 1.0.2 [8]
- Solvent Correction: Calculating solvent-corrected electronic energies using the Extended Conductor-like Polarizable Continuum Solvation Model (CPCM-X) [8]
- Energy Difference Calculation: Computing reduction potential as the difference between electronic energies of non-reduced and reduced structures (in volts) [8]
For electron affinity benchmarking, the protocol omitted solvent correction and compared predicted versus experimental gas-phase values for simple main-group organic/inorganic species and organometallic coordination complexes [8].
Validation of Fluid Correlation Peaks
An example from materials science demonstrates experimental validation of computational predictions for fluid systems [9]. Researchers tested the universality of correlation peaks in radial distribution functions (RDFs, g(r)) through:
- Experimental System Preparation: Creating 2D colloidal suspensions with tunable interactions using rotating electric fields [9]
- Microscopy and Tracking: Using optical microscopy to track individual particle positions over time [9]
- RDF Calculation: Computing experimental g(r) from particle position data [9]
- Peak Analysis: Decomposing g(r) into correlation peaks pₐ(r) representing different coordination spheres using Voronoi decomposition and shortest path graph concepts [9]
- Comparison with Simulation: Comparing experimentally derived peak parameters (norm, mean distance, mean square displacement) with molecular dynamics simulations of 2D and 3D systems [9]
Quantitative Benchmarking Results: A Case Study
The benchmarking study of OMol25-trained NNPs provides illustrative quantitative results comparing multiple methods across different chemical systems [8]. The table below summarizes performance metrics for reduction potential prediction:
Table 1: Performance of Computational Methods for Predicting Reduction Potentials [8]
| Method | Set | MAE (V) | RMSE (V) | R² |
|---|---|---|---|---|
| B97-3c | OROP | 0.260 (0.018) | 0.366 (0.026) | 0.943 (0.009) |
| B97-3c | OMROP | 0.414 (0.029) | 0.520 (0.033) | 0.800 (0.033) |
| GFN2-xTB | OROP | 0.303 (0.019) | 0.407 (0.030) | 0.940 (0.007) |
| GFN2-xTB | OMROP | 0.733 (0.054) | 0.938 (0.061) | 0.528 (0.057) |
| eSEN-S | OROP | 0.505 (0.100) | 1.488 (0.271) | 0.477 (0.117) |
| eSEN-S | OMROP | 0.312 (0.029) | 0.446 (0.049) | 0.845 (0.040) |
| UMA-S | OROP | 0.261 (0.039) | 0.596 (0.203) | 0.878 (0.071) |
| UMA-S | OMROP | 0.262 (0.024) | 0.375 (0.048) | 0.896 (0.031) |
| UMA-M | OROP | 0.407 (0.082) | 1.216 (0.271) | 0.596 (0.124) |
| UMA-M | OMROP | 0.365 (0.038) | 0.560 (0.064) | 0.775 (0.053) |
For electron affinity prediction, the study reported these results:
Table 2: Performance of Computational Methods for Predicting Electron Affinities (Main-Group Species) [8]
| Method | MAE (eV) | RMSE (eV) | R² |
|---|---|---|---|
| r2SCAN-3c | 0.130 | 0.176 | 0.984 |
| ωB97X-3c | 0.154 | 0.206 | 0.977 |
| g-xTB | 0.222 | 0.279 | 0.958 |
| GFN2-xTB | 0.236 | 0.313 | 0.947 |
| eSEN-S | 0.527 | 0.664 | 0.763 |
| UMA-S | 0.189 | 0.256 | 0.965 |
| UMA-M | 0.349 | 0.453 | 0.889 |
These quantitative results reveal several important patterns. First, performance varies significantly across different chemical domains (main-group vs. organometallic species). Second, no single method outperforms all others across all metrics and systems, highlighting the importance of context-dependent method selection. Third, despite not explicitly incorporating charge-based physics, some NNPs (particularly UMA-S) achieve accuracy competitive with traditional computational methods [8].
Table 3: Key Research Reagent Solutions for Computational Benchmarking
| Resource Category | Specific Examples | Function and Purpose |
|---|---|---|
| Reference Datasets | OMol25 dataset [6], Experimental reduction potential data [8], Experimental electron affinity data [8] | Provide high-quality reference data for training and benchmarking computational models |
| Benchmarking Frameworks | MLIPAudit [7], CASP challenges [4] | Standardized platforms for evaluating and comparing model performance |
| Experimental Validation Systems | 2D colloidal suspensions with tunable interactions [9] | Enable direct experimental testing of computational predictions under controlled conditions |
| Software Tools | geomeTRIC [8], CPCM-X solvation model [8], LAMMPS [9] | Implement geometry optimization, solvation corrections, and molecular dynamics simulations |
| Statistical Analysis Methods | Mean Absolute Error, Root Mean Square Error, R² [8] [1] | Quantify model performance and enable objective comparisons |
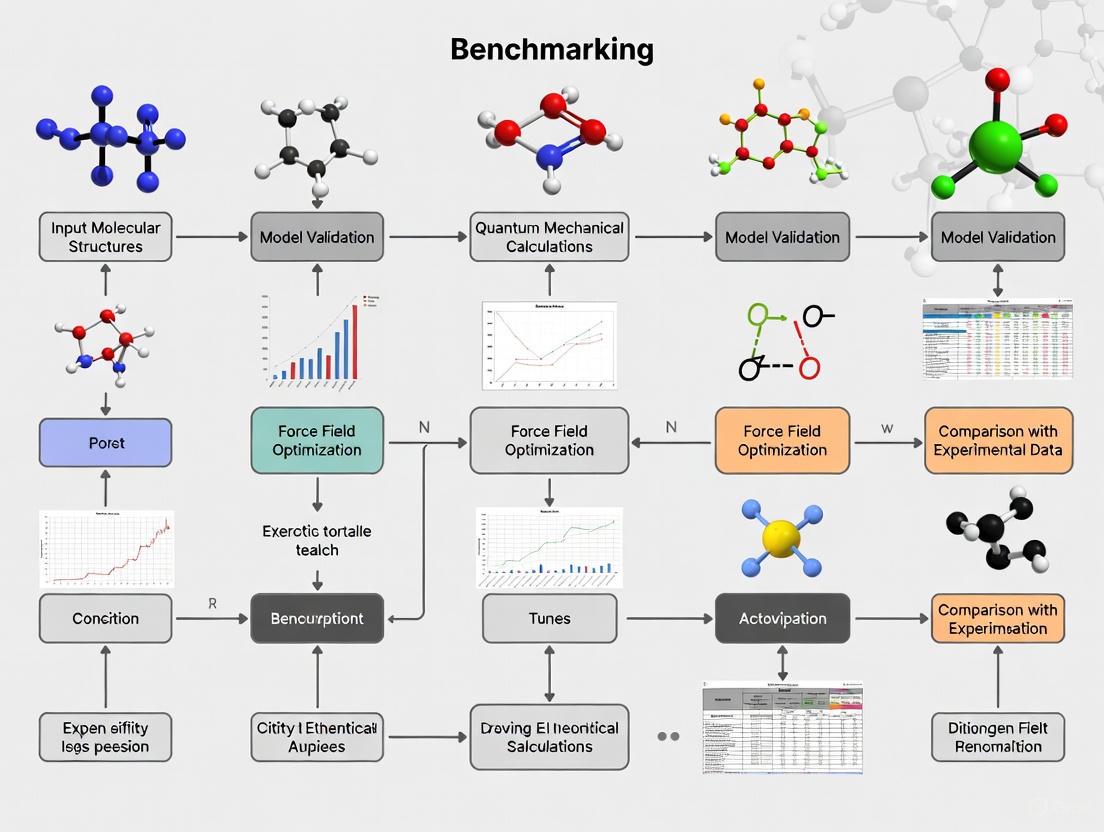
Workflow Diagram of the Benchmarking Process
The following diagram visualizes the systematic workflow for benchmarking computational models against experimental data:
Diagram 1: Benchmarking Process Workflow
Current Challenges and Future Directions
Despite methodological advances, benchmarking in computational chemistry faces persistent challenges. Data quality and availability remain significant constraints, particularly for systems requiring complex experimental measurements [4]. Overlap between training and evaluation datasets can lead to overoptimistic performance estimates, while structurally complex or flexible binding sites present particular difficulties for methods like molecular docking [4]. The field also grapples with establishing standardized evaluation protocols that balance comprehensiveness with practical feasibility [7].
Future progress requires addressing several critical needs. There is a growing consensus for developing diverse, high-quality datasets that reflect real-world applications rather than idealized systems [4]. The community would benefit from blinded evaluation methods to reduce unconscious bias, and continuous benchmarking platforms that track performance improvements over time [4]. As noted in drug discovery, "unlike protein structure prediction, which has been continually improved through CASP for over 30 years, the small molecule drug discovery community lacks equivalent, sustained frameworks for progress" [4].
Emerging approaches show promise for addressing these challenges. Community-driven benchmarking initiatives like MLIPAudit create shared reference points for assessing model accuracy, robustness, and generalization [7]. Multi-fidelity benchmarks that incorporate both high-level theoretical reference data and experimental measurements can provide more comprehensive validation [8]. Uncertainty quantification is increasingly recognized as essential for establishing predictive credibility, moving beyond point estimates to probabilistic predictions that acknowledge methodological limitations [3].
Benchmarking constitutes the essential process that connects computational predictions with experimental reality in chemistry. Through systematic comparison against reliable reference data, benchmarking transforms abstract computational methods into validated tools for scientific discovery and application. The process requires careful design—from defining clear objectives and selecting appropriate methods to choosing representative datasets and meaningful performance metrics.
As computational methods grow increasingly complex, particularly with the rise of machine learning approaches, rigorous benchmarking becomes ever more critical. It provides the evidentiary foundation needed to establish trust in computational predictions, especially when those predictions inform high-stakes decisions in drug development, materials design, or safety assessment. By identifying both strengths and limitations of computational approaches, benchmarking not only guides current method selection but also illuminates the path for future methodological improvements. In this way, benchmarking serves as both the quality control mechanism for existing methods and the innovation engine driving computational chemistry forward.
In the rapidly advancing field of computational chemistry, benchmarking has emerged as the cornerstone of scientific progress and validation. As large atomistic models (LAMs) and complex computational methods transform drug discovery and materials science, rigorous benchmarking provides the essential framework for distinguishing genuine advancements from mere algorithmic artifacts. Benchmarking serves as the critical evaluation mechanism that ensures computational tools meet the stringent requirements of scientific accuracy, reliability, and reproducibility before they can be trusted in real-world applications such as drug design and material development [10] [4].
The fundamental importance of benchmarking stems from its role in bridging the gap between theoretical development and practical application. Unlike fields where validation is straightforward, computational chemistry deals with complex molecular systems where even minor errors in energy calculations—on the scale of 1 kcal/mol—can lead to erroneous conclusions about molecular stability or binding affinity [11]. As noted in recent assessments of the field, the lack of sustained, community-wide benchmarking efforts has significantly impeded progress in critical areas like binding pose and activity prediction, where only 26-46% of ligands can be accurately regenerated within experimental uncertainty using current methods [4].
This whitepaper establishes a comprehensive framework for understanding why benchmarking is non-negotiable in computational chemistry research. By examining current benchmarking methodologies, analyzing performance data across domains, and providing practical implementation protocols, we demonstrate how systematic evaluation accelerates scientific discovery while preventing costly missteps in downstream applications.
The Benchmarking Imperative: Addressing Critical Gaps
The Universality Challenge in Atomistic Modeling
The pursuit of universal potential energy surfaces (PES) represents one of the most ambitious goals in computational chemistry, yet benchmarking reveals significant gaps between current capabilities and this ideal. Recent analyses through the LAMBench framework demonstrate that even state-of-the-art large atomistic models (LAMs) struggle with true universality across diverse chemical domains [10]. These models exhibit dramatically variable performance when applied across different research domains, particularly when trained on domain-specific data such as the MPtrj dataset for inorganic materials (using PBE/PBE+U functionals) versus small molecules requiring higher-level ωB97M functionals [10].
The fundamental challenge lies in the inherent incompatibilities between data generated across different computational chemistry domains. Variations in exchange-correlation functionals, basis sets, and pseudopotentials create systematic discrepancies that prevent seamless integration of training data [10]. This fragmentation directly impedes the development of truly universal models, as evidenced by benchmarking results showing that models excelling in one domain (e.g., inorganic materials) often underperform in others (e.g., biomolecular systems) [10].
The Reproducibility Crisis in Predictive Modeling
Beyond accuracy metrics, benchmarking reveals critical limitations in model reproducibility and stability—factors essential for reliable scientific application. In clinical diagnostic applications, large language models exhibit concerning variability, generating different responses even when input prompts, model architecture, and parameters remain identical [12]. This inconsistency poses substantial risks in diagnostic settings where the same patient case might yield divergent suggestions, potentially undermining clinical decision-making [12].
Similar challenges manifest in molecular dynamics simulations, where non-conservative models—those predicting forces directly rather than deriving them from energy gradients—can demonstrate high apparent accuracy in static evaluations yet prove unstable in actual simulations [10] [13]. The LAMBench evaluations systematically document this phenomenon, showing that models failing conservativeness requirements generate unreliable molecular dynamics trajectories despite excellent performance on energy prediction benchmarks [10].
Table 1: Performance Variability of Computational Models Across Domains
| Model Category | Primary Domain | Transfer Performance | Critical Limitations |
|---|---|---|---|
| Domain-Specific LAMs (MACE-MP-0, SevenNet-0) | Inorganic Materials | Poor transfer to biomolecular systems | Trained on PBE/PBE+U level data incompatible with chemical accuracy requirements |
| Small Molecule LAMs (AIMNet, Nutmeg) | Organic/Small Molecules | Limited transfer to materials science | Requires hybrid functionals (ωB97M) not used in materials science |
| Universal Models (UMA, eSEN) | Multiple Domains | Moderate cross-domain performance | Performance variations across chemical spaces; computational expense |
| Clinical LLMs | Medical Diagnostics | Variable across clinical specialties | Output variability even with identical inputs |
The Applicability Domain Problem
Benchmarking consistently reveals that models perform substantially worse on out-of-distribution examples compared to their advertised capabilities on in-distribution test sets. This applicability domain problem particularly impacts real-world deployment where models encounter chemical spaces not represented in their training data [14]. Comprehensive benchmarking of quantitative structure-activity relationship (QSAR) models for toxicokinetic and physicochemical properties demonstrates that prediction accuracy decreases markedly when compounds fall outside the model's defined applicability domain [14].
The consequences of this limitation are particularly significant in drug discovery, where activity cliffs—cases where small structural changes cause dramatic affinity differences—often prove most valuable for optimization yet represent precisely the scenarios where models frequently fail [4]. Without rigorous benchmarking that specifically tests these edge cases, models may appear deceptively competent while failing in the most critical applications.
Current Benchmarking Frameworks and Performance Metrics
Established Benchmarking Systems
The computational chemistry community has developed several specialized benchmarking frameworks to address distinct evaluation needs. The table below summarizes key frameworks and their primary applications:
Table 2: Specialized Benchmarking Frameworks in Computational Chemistry
| Benchmark Framework | Primary Focus | Key Metrics | Domain Coverage |
|---|---|---|---|
| LAMBench [10] | Large Atomistic Models (LAMs) | Generalizability, Adaptability, Applicability | Broad coverage across materials, molecules, and biomolecules |
| QUID [11] | Ligand-Pocket Interactions | Interaction energy accuracy, Force prediction | Non-covalent interactions in biological systems |
| MOFSimBench [13] | Metal-Organic Frameworks | Structure optimization, Molecular dynamics stability, Host-guest interactions | Porous materials for catalysis and storage |
| ADMET Benchmarking [14] | Toxicokinetic Properties | Regression R², Balanced accuracy, Applicability domain adherence | Drug-like molecules and industrial chemicals |
Quantitative Performance Comparisons
Rigorous benchmarking provides crucial quantitative comparisons between computational methods. Recent evaluations of machine learning interatomic potentials (MLIPs) on MOFSimBench reveal significant performance variations across different simulation tasks [13]:
Table 3: MLIP Performance on MOFSimBench Tasks (100 structures)
| Model | Structure Optimization (<10% volume change) | MD Stability (<10% volume change) | Bulk Modulus MAE | Host-Guest Interaction MAE |
|---|---|---|---|---|
| PFP v8.0.0 | 92/100 | 89/100 | 1.98 GPa | 0.029 eV |
| eSEN-OAM | 88/100 | 91/100 | 1.52 GPa | 0.031 eV |
| orb-v3-omat+D3 | 87/100 | 88/100 | 2.15 GPa | 0.035 eV |
| uma-s-1p1 | 86/100 | Not tested | 2.01 GPa | 0.033 eV |
For ligand-pocket interactions, the QUID benchmark establishes a "platinum standard" through agreement between complementary coupled cluster (CC) and quantum Monte Carlo (QMC) methods, achieving remarkable interaction energy agreement of 0.5 kcal/mol [11]. This high-accuracy benchmark reveals that while several dispersion-inclusive density functional approximations provide reasonable energy predictions, their atomic van der Waals forces often differ substantially in magnitude and orientation [11]. Meanwhile, semiempirical methods and empirical force fields require significant improvements in capturing non-covalent interactions, particularly for out-of-equilibrium geometries [11].
In ADMET prediction, comprehensive benchmarking of twelve QSAR tools shows that models for physicochemical properties (average R² = 0.717) generally outperform those for toxicokinetic properties (average R² = 0.639 for regression, average balanced accuracy = 0.780 for classification) [14]. This performance gap highlights the greater complexity of biological interactions compared to pure compound characteristics.
Experimental Protocols for Rigorous Benchmarking
Protocol 1: Evaluating Model Generalizability
The LAMBench framework provides a systematic methodology for assessing model generalizability across three critical dimensions: in-distribution performance, out-of-distribution performance, and cross-domain transfer capability [10].
Procedure:
- Dataset Curation: Compile diverse test sets representing distinct chemical domains (biomolecules, electrolytes, metal complexes, materials)
- In-Distribution Testing: Evaluate performance on test sets randomly split from training data distribution
- Out-of-Distribution Testing: Assess performance on chemically distinct systems not represented in training data
- Cross-Domain Transfer: Measure performance when models trained in one domain (e.g., materials) are applied to another (e.g., biomolecules)
- Statistical Analysis: Compute metrics including Mean Absolute Error (MAE), Root Mean Square Error (RMSE), and failure rate analysis
Key Considerations: Domain-specific functional preferences create inherent incompatibilities; for example, materials science typically employs PBE/PBE+U functionals while chemical accuracy requires hybrid functionals like ωB97M [10]. Benchmarking must therefore account for these fundamental methodological differences when assessing cross-domain performance.
Protocol 2: Assessing Reproducibility and Repeatability
For clinical and diagnostic applications, a specialized statistical framework quantifies both repeatability (agreement under identical conditions) and reproducibility (agreement under different, pre-specified conditions) [12].
Procedure:
- Experimental Setup: Select multiple models (commercial and open-source) representing different architectures and scales
- Prompt Variation: Utilize multiple validated prompts designed to elicit distinct reasoning strategies (chain-of-thought, differential diagnosis, intuitive, analytic, and Bayesian reasoning)
- Repeated Sampling: Execute numerous independent runs (R=100) per prompt-case-model combination
- Semantic Consistency Assessment: Measure stability in output meaning across repeated runs using embedding-based similarity metrics
- Internal Consistency Quantification: Assess token-level generation variability across repetitions
Validation Datasets: Employ both standardized benchmarks (e.g., MedQA with 518 USMLE-style questions) and real-world challenging cases (e.g., 90 rare disease cases from Undiagnosed Diseases Network) [12].
Protocol 3: Validating Practical Applicability
The MOFSimBench framework provides a comprehensive protocol for evaluating practical model performance on real-world simulation tasks [13].
Procedure:
- Structure Optimization Task
- Input: 100 diverse MOF/COF/zeolite structures
- Method: Full geometry optimization using target MLIP
- Metric: Volume change percentage compared to DFT-optimized reference structures
- Success Criterion: |ΔV| < 10% compared to DFT reference
Molecular Dynamics Stability Assessment
- Input: 100 structures after optimization
- Method: NPT simulation (50 ps, 300K, 1 bar) after equilibration
- Metric: Volume change between initial and final structures
- Success Criterion: |ΔV| < 10% during production MD
Bulk Property Prediction
- Input: Strain series for each optimized structure
- Method: Birch-Murnaghan equation of state fitting
- Metric: Mean Absolute Error (MAE) for bulk modulus compared to DFT
- Quality Control: Exclude structures where fitted minimum volume deviates >1% from optimized volume
Host-Guest Interaction Accuracy
- Input: 26 MOF structures with CO₂/H₂O adsorbates at multiple interaction distances
- Method: Single-point energy and force calculations
- Metric: MAE for interaction energies and forces compared to DFT reference across repulsion, equilibrium, and weak-attraction regimes
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 4: Essential Benchmarking Tools and Resources
| Tool/Resource | Type | Primary Function | Key Applications |
|---|---|---|---|
| LAMBench [10] | Benchmarking System | Evaluating Large Atomistic Models | Generalizability, adaptability, and applicability assessment |
| QUID Framework [11] | Quantum-Chemical Benchmark | Platinum-standard interaction energies | Ligand-pocket non-covalent interactions |
| MOFSimBench [13] | Specialized Benchmark | MLIP evaluation for porous materials | MOF structure, stability, and host-guest properties |
| OPERa [14] | QSAR Toolsuite | Predicting physicochemical properties | ADMET profiling and chemical safety assessment |
| torch-dftd [13] | Dispersion Correction | Adding dispersion forces to MLIPs | Accurate non-covalent interaction modeling |
| RDKit [14] | Cheminformatics | Chemical structure standardization | Data curation and descriptor calculation |
| PBE0+MBD [11] | Density Functional | Reference quantum calculations | Generating high-quality training and benchmark data |
Implementation Framework: Integrating Benchmarking into Research Workflows
Establishing a Continuous Benchmarking Culture
The most effective benchmarking extends beyond periodic validation to become an integral part of the research lifecycle. This requires adopting several key practices:
Pre-registration of Benchmarking Protocols: Before model development begins, researchers should pre-register their intended benchmarking strategies, including datasets, evaluation metrics, and comparison baselines. This approach prevents retrospective benchmark selection that potentially inflates perceived performance [4].
Blinded Evaluation Methods: Following the successful model of the Critical Assessment of Structure Prediction (CASP) in protein folding, small molecule drug discovery should implement blinded evaluations using unreleased experimental data to prevent unconscious optimization toward known results [4].
Multi-dimensional Performance Tracking: Rather than relying on single metrics, comprehensive benchmarking should simultaneously track accuracy, computational efficiency, robustness to input variations, and failure modes across diverse chemical spaces [10] [13].
Community-Wide Benchmarking Initiatives
Individual efforts alone cannot address the systemic benchmarking challenges in computational chemistry. The field requires coordinated community initiatives:
Standardized Dataset Generation: Following the example of the OMol25 dataset—which comprises over 100 million quantum chemical calculations requiring 6 billion CPU-hours—the community should prioritize creating shared, high-accuracy datasets spanning diverse chemical domains [15].
Open Leaderboards and Transparent Reporting: Initiatives like the interactive LAMBench leaderboard provide ongoing community assessment of model capabilities, enabling researchers to identify strengths and limitations before applying models to specific research problems [10].
Cross-disciplinary Benchmarking Consortia: Successful benchmarking requires integration across traditionally separate domains. The collaboration between quantum chemists, materials scientists, and pharmaceutical researchers in developing QUID demonstrates the power of cross-domain collaboration in establishing meaningful benchmarks [11].
Benchmarking represents a non-negotiable foundation for reliable computational chemistry research. As the field progresses toward increasingly complex models and applications, systematic evaluation becomes ever more critical for distinguishing genuine advances from methodological artifacts. The frameworks, protocols, and resources outlined in this whitepaper provide a roadmap for integrating rigorous benchmarking throughout the research lifecycle.
The evidence is clear: without comprehensive benchmarking, computational chemistry risks generating elegant but unreliable models that fail in critical applications. From drug discovery to materials design, the consequences of unvalidated models include wasted resources, missed opportunities, and ultimately, erosion of trust in computational methods. By embracing the benchmarking imperative, the research community can accelerate genuine progress while ensuring the reliability and reproducibility that form the bedrock of scientific integrity.
As the field stands at what many call "an AlphaFold moment" for atomistic simulation [15], the establishment of robust, community-wide benchmarking practices will determine whether this promise translates into genuine scientific advancement or remains an unfulfilled potential. The tools, frameworks, and methodologies now exist to make comprehensive benchmarking routine rather than exceptional—the responsibility lies with the research community to implement them consistently and rigorously.
In computational chemistry and drug discovery, benchmarking is the systematic process of evaluating and comparing the performance of predictive models against standardized datasets and metrics. This process is fundamental for assessing model robustness, reliability, and practical utility in real-world applications such as toxicity prediction and molecular property estimation [14]. The reliability of any computational model is intrinsically linked to three interconnected concepts: the Training Set, which provides the foundational data for model building; the Applicability Domain (AD), which defines the chemical space where model predictions are reliable; and Validation Metrics, which quantitatively measure model performance [16]. Together, these components form a critical framework for establishing confidence in computational predictions, guiding researchers in identifying the most suitable tools for chemical safety assessment, drug discovery, and material design [14].
Core Terminology Deep Dive
Applicability Domain (AD)
The Applicability Domain (AD) represents the "response and chemical structure space in which the model makes predictions with a given reliability" [17]. It establishes the boundaries within which a model can be confidently applied, based on the chemical space covered by its training data. Predictions for molecules falling outside the AD are considered unreliable [17].
Key Methods for Defining AD:
- Novelty Detection: Identifies compounds dissimilar to training set molecules using only their explanatory variables, without considering the underlying classifier or class labels [17].
- Confidence Estimation: Uses information from the trained classifier itself, typically measuring the distance of a compound to the model's decision boundary [17].
- Subgroup Discovery (SGD): Identifies domains of applicability as logical conjunctions of simple conditions (e.g., on lattice vectors or bond distances) where model error is substantially lower than its global average [18].
Training Sets
Training sets consist of chemical compounds with known experimental or calculated properties used to build predictive models. The composition and quality of training data directly influence model performance and the extent of its applicability domain [14].
Critical Aspects of Training Set Construction:
- Data Curation: Requires standardization of chemical structures, removal of duplicates, handling of inorganic and organometallic compounds, neutralization of salts, and identification of outliers [14].
- Chemical Space Analysis: Involves mapping training compounds against reference chemical spaces (e.g., approved drugs, industrial chemicals, natural products) to understand coverage limitations [14].
- Assessing Modelability: Indexes such as the Rivality Index (RI) can predict dataset suitability for modeling before model building, with high positive RI values indicating compounds outside the AD [16].
Validation Metrics
Validation metrics provide quantitative measures of model performance and prediction reliability, enabling comparison between different modeling approaches [14] [19].
Classification Metrics:
- Area Under the Receiver Operating Characteristic Curve (AUC-ROC): Measures the trade-off between true positive and false positive rates across different classification thresholds [19].
- Area Under the Precision-Recall Curve (AUC-PR): Particularly valuable for imbalanced datasets where inactive compounds significantly outnumber actives [19].
- Balanced Accuracy: Appropriate for balanced sets, calculated as the average of sensitivity and specificity [16].
Regression Metrics:
- Coefficient of Determination (R²): Measures the proportion of variance in the response variable explained by the model [14].
- Mean Absolute Error (MAE): Average magnitude of errors between predicted and experimental values [1].
- Root Mean Square Error (RMSE): Places higher weight on larger errors due to squaring of differences [1].
Uncertainty Quantification Metrics:
- Spearman's Rank Correlation: Assesses how well uncertainty estimates rank the observed errors [20].
- Negative Log Likelihood (NLL): Measures how well the uncertainty distribution explains the observed errors [20].
- Error-Based Calibration: The superior metric that checks if the average absolute error or RMSE matches the predicted uncertainty across uncertainty ranges [20].
Experimental Protocols for Benchmarking
Comprehensive Model Benchmarking Framework
Table 1: Key Steps in Model Benchmarking
| Step | Protocol Description | Purpose |
|---|---|---|
| Dataset Collection | Gather experimental data from literature and databases (e.g., ChEMBL, PHYSPROP) using systematic search terms and API access [14]. | Ensures comprehensive coverage of chemical space and endpoints. |
| Data Curation | Standardize structures, remove duplicates, neutralize salts, identify outliers using Z-scores, and resolve inconsistent values across datasets [14]. | Improves data quality and reliability for model training and validation. |
| Chemical Space Analysis | Plot datasets against reference chemical spaces (approved drugs, industrial chemicals) using molecular fingerprints and PCA [14]. | Determines chemical categories covered and identifies potential biases. |
| Model Training | Implement multiple algorithms (RF, SVM, Neural Networks) with appropriate hyperparameter settings and validation techniques [17]. | Enables fair comparison of different modeling approaches. |
| Performance Evaluation | Assess models using multiple metrics (AUC-ROC, R², etc.) with emphasis on performance inside the applicability domain [14]. | Provides comprehensive assessment of model strengths and limitations. |
Establishing the Applicability Domain
Table 2: Methods for Defining Applicability Domain
| Method Category | Specific Techniques | Implementation Considerations |
|---|---|---|
| Novelty Detection | Leverage, vicinity, distance to training set centroid, k-NN similarity [14] [17]. | Does not use class label information; based solely on feature space proximity. |
| Confidence Estimation | Class probability estimates, distance to decision boundary, classifier stability [17]. | Uses information from the trained classifier; generally more powerful than novelty detection. |
| Hybrid Approaches | ADAN (6 measurements), Consensus models, Random Forest with v-NN [16]. | Combines multiple approaches; often provides systematically better performance. |
Figure 1: Workflow for Comprehensive Model Benchmarking in Computational Chemistry
Quantitative Benchmarks and Performance Standards
Performance Across Chemical Properties
Table 3: Benchmarking Results for Property Prediction Models
| Property Type | Best Performing Models | Performance Metrics | Chemical Space Coverage |
|---|---|---|---|
| Physicochemical (PC) Properties | OPERA, Random Forests | R² average = 0.717 [14] | Drugs, industrial chemicals, pesticides [14] |
| Toxicokinetic (TK) Properties | Ensemble methods, SVM | R² average = 0.639 (regression)\nBalanced accuracy = 0.780 (classification) [14] | Relevant for ADMET profiling [14] |
| Bioactivity Prediction | Deep Learning, SVM, Random Forests | AUC-ROC: 0.8-0.9 range [19] | Diverse targets from ChEMBL (1300+ assays) [19] |
Uncertainty Quantification Benchmarking
Performance evaluation of uncertainty quantification methods requires specific metrics that differ from standard model validation [20]. The error-based calibration approach introduced by Levi et al. has been shown superior to metrics like Spearman's rank correlation, miscalibration area, and negative log likelihood [20]. This method validates that the relationship between predicted uncertainties and observed errors follows the expected statistical behavior where the average absolute error should approximate $\sqrt{\frac{2}{\pi}}\sigma$ and the root mean square error should approximate $\sigma$ for a suitably large subset of predictions [20].
Figure 2: Applicability Domain Assessment Workflow for Individual Predictions
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Tools for Computational Chemistry Benchmarking
| Tool Category | Specific Tools | Function and Application |
|---|---|---|
| Chemical Databases | ChEMBL, PubChem, DrugBank, PHYSPROP | Sources of experimental data for training and validation [14] |
| Descriptor Calculation | RDKit, CDK, jCompoundMapper | Generation of molecular fingerprints and descriptors [14] |
| Modeling Algorithms | Random Forest, SVM, Neural Networks, k-NN | Core algorithms for building predictive models [17] |
| Validation Metrics | AUC-ROC, AUC-PR, R², Balanced Accuracy | Quantitative assessment of model performance [19] |
| Uncertainty Quantification | Ensemble methods, Latent Space Distance, Evidential Regression | Estimating prediction reliability and confidence intervals [20] |
Robust benchmarking in computational chemistry requires meticulous attention to training set composition, clear definition of applicability domains, and appropriate selection of validation metrics. The integration of these three components forms the foundation for developing reliable predictive models that can accelerate drug discovery and chemical safety assessment. Current research indicates that while no single algorithm universally outperforms others across all chemical domains, systematic benchmarking enables identification of optimal approaches for specific prediction tasks [14] [19]. Future methodological improvements should focus on developing more sophisticated applicability domain definitions, standardized benchmarking protocols, and enhanced uncertainty quantification techniques to further increase confidence in computational predictions.
The Role of Benchmarking in New Approach Methodologies (NAMs) and Reducing Animal Testing
The pharmaceutical and regulatory landscape is undergoing a fundamental transformation, marked by a strategic shift away from traditional animal testing toward human-relevant New Approach Methodologies (NAMs). This transition, championed by regulatory bodies including the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), leverages advanced computational models, in vitro systems, and AI-driven analytics to evaluate drug safety and efficacy [21] [22] [23]. Benchmarking serves as the critical bridge connecting innovative computational methodologies to regulatory acceptance and real-world application. By providing a rigorous, standardized framework for validation, benchmarking ensures that computational models in chemistry and biology are predictive, reliable, and trustworthy enough to inform high-stakes decisions in drug development, thereby accelerating the reduction of animal use in accordance with the 3Rs principles (Replace, Reduce, Refine) [22] [23].
The Critical Function of Benchmarking in Computational NAMs
Within computational chemistry and drug discovery, benchmarking is not merely a performance check; it is the foundational process that establishes scientific validity and regulatory confidence. It systematically answers the question: "Can this model reliably recapitulate or predict complex biological and chemical phenomena for its intended purpose?"
Fragmented, domain-specific benchmarks have historically impeded progress toward universal models. The LAMBench benchmark, for instance, was created to address the lack of comprehensive evaluation frameworks for Large Atomistic Models (LAMs) that aim to approximate a universal potential energy surface [10]. It assesses models on three core capabilities:
- Generalizability: Accuracy across diverse, unseen atomistic systems.
- Adaptability: Capacity to be fine-tuned for tasks beyond primary training, such as structure-property relationships.
- Applicability: Stability and efficiency in real-world simulations, like molecular dynamics [10].
Similarly, in the field of binding prediction, Kramer et al. highlight the lack of sustained community benchmarks akin to the CASP challenge for protein structure prediction. This gap hinders the reliable comparison of methods for predicting ligand binding poses and affinities, which is a cornerstone of structure-based drug design [4].
The Critical Gap in Pose and Activity Prediction
The absence of long-term, blinded community benchmarks for binding pose- and activity prediction (P-AP) has significantly hampered progress in computational drug discovery. Unlike the protein structure prediction field, which has been rigorously advanced through the decades-long CASP challenge, P-AP lacks an equivalent framework. This makes it difficult for researchers to compare methods and track genuine improvements, ultimately limiting the adoption of reliable computational tools in the drug discovery pipeline [4].
Quantitative Benchmarking of Computational Chemistry Models
Robust benchmarking requires standardized datasets and protocols to objectively compare the performance of different computational methods. The following examples illustrate how this is practiced in the field.
Benchmarking Neural Network Potentials on Charge-Related Properties
A key study benchmarked Neural Network Potentials (NNPs) trained on Meta's Open Molecules 2025 (OMol25) dataset against experimental data for reduction potential and electron affinity. The results were compared to traditional low-cost computational methods, revealing the strengths and weaknesses of data-driven NNPs, even when they do not explicitly model charge-based physics [8].
Table 1: Performance of Computational Methods in Predicting Experimental Reduction Potentials (Mean Absolute Error, V)
| Method | Main-Group Set (OROP) | Organometallic Set (OMROP) |
|---|---|---|
| B97-3c (DFT) | 0.260 | 0.414 |
| GFN2-xTB (SQM) | 0.303 | 0.733 |
| eSEN-S (OMol25 NNP) | 0.505 | 0.312 |
| UMA-S (OMol25 NNP) | 0.261 | 0.262 |
| UMA-M (OMol25 NNP) | 0.407 | 0.365 |
Source: Adapted from [8]. MAE values in Volts (V). Lower is better.
Benchmarking on Non-Equilibrium Structures
The Wiggle150 benchmark addresses a critical gap by focusing on highly strained, non-equilibrium molecular conformations. This is essential for validating models used in ab initio molecular dynamics and reaction-path exploration, where molecules frequently adopt geometries far from their equilibrium states. The benchmark comprises 150 strained conformations of three molecules (adenosine, benzylpenicillin, and efavirenz), with reference energies derived from high-level DLPNO-CCSD(T)/CBS calculations. In this challenging test, the neural network potential AIMNet2 was identified as particularly robust among the methods surveyed [24].
Experimental Protocol for Benchmarking Reduction Potentials
The methodology for benchmarking computational models against experimental reduction potentials, as detailed in [8], involves a precise multi-step workflow:
- Structure Optimization: The non-reduced and reduced structures of each species are optimized using the NNP (or other method) in question, typically employing a geometry optimization library like
geomeTRIC. - Solvent Correction: Each optimized structure is processed through an implicit solvation model, such as the Extended Conductor-like Polarizable Continuum Model (CPCM-X), to obtain a solvent-corrected electronic energy.
- Energy Difference Calculation: The predicted reduction potential is calculated as the difference in electronic energy (in electronvolts) between the non-reduced and reduced structures. This value is directly comparable to the experimental reduction potential in volts.
- Statistical Comparison: The predicted values are compared against the experimental dataset. Standard metrics like Mean Absolute Error (MAE), Root Mean Squared Error (RMSE), and the coefficient of determination (R²) are computed to quantify accuracy.
A Framework for Generating High-Quality Negative Data
A major challenge in validating virtual high-throughput screening (vHTS) pipelines is the lack of high-quality negative data (i.e., confirmed non-binders). An innovative computational approach generates such data without additional experiments:
- Ligand Randomization: Docking known ligands into unrelated protein structures from the Protein Data Bank (PDB) to create a set of confirmed non-binders.
- Isomer Generation: Creating structural isomers of known active ligands, which are highly likely to be inactive, providing negative data points that are closely matched in molecular properties to the positives.
This method produces practically unlimited negative data that is superior in quality and quantity to previously available sets, enabling rigorous validation of every step in a vHTS pipeline to ensure it adds genuine enrichment over random selection [25].
The Path to Regulatory Acceptance: A Tiered Workflow
For a NAM to be adopted in regulatory decision-making, it must undergo a rigorous evaluation process with regulatory agencies. The following workflow outlines the key stages and interactions for achieving regulatory acceptance.
Diagram 1: Path to Regulatory Acceptance of NAMs. The workflow illustrates the iterative stages of engagement with regulators like the EMA, from initial briefing to full qualification. ITF: Innovation Task Force. Adapted from [23].
The context of use is a formal description of the specific circumstances under which the NAM is applied and is the cornerstone of this process. Regulatory requirements are most stringent for NAMs intended to replace animal studies in safety assessment, where a formal qualification opinion from the Committee for Human Medicinal Products (CHMP) may be required. For applications in primary pharmacology or proof-of-concept, acceptance can be achieved on a case-by-case basis within a marketing authorization application [23].
The successful implementation and benchmarking of NAMs rely on a suite of computational and experimental tools.
Table 2: Key Research Reagent Solutions for NAMs Implementation
| Tool / Resource | Type | Function in NAMs & Benchmarking |
|---|---|---|
| Organoids & 3D Cell Cultures | In Vitro System | Provides complex, human-relevant tissue models for efficacy and toxicity testing, bridging the gap between 2D cells and in vivo models [22]. |
| Organs-on-a-Chip | In Vitro System | Microphysiological systems that mimic human organ function and interaction for high-fidelity safety and ADME profiling [21] [22]. |
| OMol25 Dataset | Computational Data | A massive dataset of >100M quantum chemical calculations used to pre-train universal neural network potentials for molecular modeling [8]. |
| LAMBench | Benchmarking Software | A comprehensive benchmarking system to evaluate the generalizability, adaptability, and applicability of Large Atomistic Models [10]. |
| Wiggle150 | Benchmarking Dataset | A curated set of 150 highly strained molecular conformations with reference energies for testing model robustness on non-equilibrium structures [24]. |
| ChemBench | Benchmarking Framework | An automated framework with >2,700 curated questions to evaluate the chemical knowledge and reasoning abilities of Large Language Models [26]. |
Benchmarking is the linchpin in the transition to a modern, human-relevant paradigm for drug safety and efficacy evaluation. Through rigorous, community-driven benchmarks like LAMBench, Wiggle150, and ChemBench, computational models achieve the validation necessary to gain the confidence of researchers and regulators. As the FDA's plan to phase out animal testing for monoclonal antibodies demonstrates, the future of drug development is inextricably linked to the continued development and validation of NAMs [21]. By adhering to structured benchmarking protocols and engaging early with regulatory pathways, the scientific community can accelerate the adoption of these innovative tools, ultimately leading to safer, more effective medicines developed through more efficient and ethical means.
Benchmarking is an indispensable practice in computational chemistry and drug development, serving as the foundational process for assessing the accuracy, reliability, and performance of computational models and experimental workflows. Within computational chemistry research, benchmarking is formally defined as the systematic process of comparing a model's predictions against reference data—whether experimental results or higher-level theoretical calculations—to establish its predictive validity and domain of applicability [27]. This process enables researchers to quantify progress, validate new methodologies, and make informed decisions based on empirical evidence rather than intuition alone.
In the broader thesis of computational model validation, benchmarking represents the critical bridge between theoretical development and practical application. It transforms abstract algorithms into trusted tools for scientific discovery and industrial application. As computational methods increasingly inform critical decisions in drug development and materials design, the rigor of benchmarking practices directly impacts the pace of innovation and the reliability of outcomes across chemical sciences and related fields [4] [28].
Critical Gaps in Current Benchmarking Practices
Methodological and Data Quality Deficiencies
Current benchmarking approaches across computational chemistry and pharmaceutical development exhibit significant shortcomings that undermine their utility and reliability.
Table 1: Primary Deficiencies in Traditional Benchmarking Approaches
| Deficiency Category | Specific Limitations | Impact on Decision-Making |
|---|---|---|
| Data Completeness | Infrequent updates failing to incorporate new data [29] | Decisions based on outdated information leading to risk underestimation |
| Data Quality | Overly broad categorization (e.g., "oncology" vs. specific cancer subtypes) [29] | Inaccurate probability of success assessments for specific targets |
| Methodological Rigor | Overly simplistic Probability of Success (POS) calculations multiplying phase transition rates [29] | Systematic overestimation of drug development success rates |
| Domain Applicability | Inadequate handling of innovative development paths (e.g., skipped phases, dual phases) [29] | Poor benchmarking for non-standard development approaches |
The pharmaceutical industry exemplifies these challenges, where traditional benchmarking often relies on static datasets that "are updated infrequently and therefore don't draw on the most up-to-date information" [29]. This temporal decay in data relevance is particularly problematic in fast-evolving fields. Furthermore, simplistic methodological approaches, such as multiplying phase transition probabilities to determine overall likelihood of success, systematically "overestimate a drug's success rate, resulting in less-than-ideal data for decision-making" [29].
Community and Infrastructure Limitations
Beyond methodological issues, the field suffers from insufficient community infrastructure and standardized practices for sustained benchmarking. Unlike protein structure prediction, which has benefited from decades of continuous community evaluation through the Critical Assessment of Structure Prediction (CASP), small molecule drug discovery "lacks equivalent, sustained frameworks for progress" [4]. This infrastructure gap manifests through several critical challenges:
- Dataset Contamination: Widespread overlap between training and evaluation datasets leads to overly optimistic performance estimates [4].
- Evaluation Complexity: Structurally complex or flexible binding sites, non-physical poses, and variable experimental data quality complicate fair comparisons [4].
- Temporal Validation Deficits: Absence of prospective, time-stamped evaluations that simulate real-world discovery scenarios [4].
- Unconfirmed Decoys: Use of unvalidated negative examples in binding affinity predictions [4].
These limitations collectively hinder reliable comparison of computational methods and obscure genuine performance improvements, ultimately slowing the translation of methodological advances into practical discovery tools.
Domain-Specific Benchmarking Challenges
Pharmaceutical Development and Clinical Translation
In drug development, benchmarking serves crucial functions in risk management, resource allocation, and regulatory strategy, yet significant gaps persist between benchmarking practices and decision-making needs. Recent empirical analyses of FDA approvals (2006-2022) reveal an average Likelihood of Approval (LoA) rate of 14.3% across leading pharmaceutical companies, with substantial variation ranging from 8% to 23% [30]. This heterogeneity underscores the limitations of one-size-fits-all benchmarking approaches.
Clinical development benchmarking extends beyond success rates to encompass operational metrics including site performance, protocol amendments, fair market value, and patient enrollment [31]. Each domain presents unique benchmarking challenges:
- Protocol Design: Benchmarking protocol amendment experiences and their impact on study duration requires "systematic assessments of the number and causes of protocol amendments" to create better, less costly protocols [31].
- Site Selection: Benchmarking first-patient-in cycle times across regions to improve site activation and trial initiation [31].
- Investigator Payments: Fair market value benchmarking to establish appropriate payments based on therapeutic area, region, and trial phase [31].
The transition between clinical phases represents a particularly critical benchmarking gap, with traditional approaches often failing to account for program-specific factors that significantly influence transition probabilities.
Computational Chemistry and Method Validation
Theoretical chemistry faces distinct benchmarking challenges rooted in the relationship between computational predictions and experimental validation. A concerning trend identified in the literature is the practice of "theory benchmarking theory," where "the quality of a model is thereby no longer measured through any relation to experiment, but purely to the similarity to another model" [27]. This self-referential approach has become so prevalent that "many manuscripts dedicated to quantum chemistry benchmarks do not feature a single experimental result" [27].
The GMTKN30 database exemplifies this issue, containing "only a small amount of experimental reference data" with "14/30 sets us[ing] as reference data estimated CCSD(T)/CBS limits" [27]. This reliance on theoretical rather than experimental benchmarks creates circular validation that may not reflect real-world predictive performance.
The recent introduction of massive datasets like Meta's Open Molecules 2025 (OMol25), containing "over 100 million quantum chemical calculations" representing "biomolecules, electrolytes, and metal complexes," offers potential improvements through unprecedented chemical diversity and data quality [15]. However, the scale of such datasets "will make training challenging for organizations without access to large numbers of GPUs," potentially creating resource-based disparities in benchmarking capabilities [15].
Table 2: Comparison of Computational Chemistry Datasets and Benchmarks
| Dataset/Benchmark | Size | Diversity | Reference Quality | Key Limitations |
|---|---|---|---|---|
| OMol25 (2025) | >100 million calculations [15] | High (biomolecules, electrolytes, metal complexes) [15] | ωB97M-V/def2-TZVPD level theory [15] | Computational resource requirements limit accessibility |
| GMTKN30 | 30 benchmark sets [27] | Moderate (main-group organic) [27] | Primarily CCSD(T)/CBS estimates [27] | Limited experimental validation; theory-only references |
| ANI Series | Millions of structures [15] | Low (simple organic, 4 elements) [15] | ωB97X/6-31G(d) level theory [15] | Limited element coverage and chemical diversity |
Experimental Protocols for Robust Benchmarking
Framework for Predictive Model Validation
Robust benchmarking requires standardized experimental protocols that ensure fair comparison and reproducible results. Based on community best practices, the following methodology provides a template for comprehensive model evaluation:
1. Dataset Curation and Validation
- Collect experimental data from diverse sources including literature, public databases (e.g., PubChem, DrugBank), and proprietary collections [14].
- Standardize chemical representations using automated procedures (e.g., RDKit Python package) to address inorganic/organometallic compounds, mixtures, and unusual elements [14].
- Resolve duplicates by averaging continuous values with standardized standard deviation <0.2; remove compounds with greater variance [14].
- Identify and exclude response outliers using Z-score analysis (Z-score >3 considered outliers) [14].
2. Chemical Space Analysis
- Establish reference chemical space using representative compounds (e.g., ECHA database for industrial chemicals, DrugBank for approved drugs) [14].
- Compute molecular descriptors (e.g., FCFP fingerprints with radius 2 folded to 1024 bits) using standardized tools [14].
- Apply Principal Component Analysis (PCA) to visualize dataset coverage relative to reference space [14].
3. Model Evaluation and Applicability Domain Assessment
- Perform external validation using temporally or structurally distinct test sets [32] [14].
- Define and apply applicability domain (AD) methods (e.g., leverage, structural vicinity) to identify reliable predictions [14].
- Employ multiple metrics including R² for regression, balanced accuracy for classification, and domain-specific measures [32] [14].
4. Performance Benchmarking
- Compare against established baselines and state-of-the-art methods [14].
- Conduct statistical significance testing on performance differences [32].
- Report performance stratified by applicability domain inclusion [14].
Community-Wide Evaluation Frameworks
Inspired by successful initiatives in structural biology, emerging frameworks for community-wide benchmarking address the need for standardized, blinded evaluations:
Temporal Splitting Protocol
- Sort drug-indication associations by approval date [32]
- Use earlier approvals for training, later approvals for testing [32]
- Simulates real-world predictive scenarios more realistically than random splits [32]
Leave-One-Out Cross-Validation
- Iteratively exclude all associations for specific indications or drug classes [32]
- Tests model performance on completely novel therapeutic areas [32]
- Particularly relevant for drug repurposing applications [32]
Stratified Performance Reporting
- Report metrics stratified by chemical classes, target families, and disease areas [14]
- Include applicability domain coverage statistics [14]
- Disclose performance variation across different data segments [14]
Essential Research Reagents and Computational Tools
The Scientist's Toolkit for Benchmarking Studies
Robust benchmarking requires both computational tools and experimental data resources. The following table catalogs essential resources identified through recent benchmarking initiatives:
Table 3: Essential Resources for Computational Chemistry Benchmarking
| Resource Category | Specific Tools/Databases | Primary Function | Key Features/Benefits |
|---|---|---|---|
| Quantum Chemical Datasets | OMol25 [15], ANI series [15], SPICE [15] | Training and validation of neural network potentials | High-accuracy calculations, diverse chemical spaces, extensive coverage |
| Drug Discovery Databases | Therapeutic Targets Database (TTD) [32], Comparative Toxicogenomics Database (CTD) [32], DrugBank [14] | Ground truth mapping for drug-indication associations | Manually curated interactions, standardized identifiers, multiple evidence levels |
| QSAR Modeling Platforms | OPERA [14], admetSAR [14] | Prediction of physicochemical and toxicokinetic properties | Applicability domain assessment, batch prediction capabilities, open-source availability |
| Cheminformatics Tools | RDKit [14], CDK (Chemistry Development Kit) [14] | Molecular standardization, descriptor calculation, fingerprint generation | Open-source, comprehensive functionality, Python integration |
| Clinical Development Data | ClinicalTrials.gov [30], internal pharmaceutical company databases [31] | Clinical trial success rates, operational metrics | Real-world development outcomes, comprehensive trial metadata |
A Roadmap for Community Action
Addressing the benchmarking gaps in computational chemistry and drug development requires coordinated community action. Based on identified challenges and emerging best practices, we propose the following roadmap:
1. Establish Sustained Benchmarking Infrastructure
- Create blinded evaluation platforms for pose and activity prediction similar to CASP [4]
- Develop mechanisms for continuous dataset updates while maintaining backward compatibility [4]
- Implement temporal validation as a standard practice rather than an exception [32]
2. Improve Dataset Quality and Diversity
- Prioritize experimental validation alongside theoretical improvements [27]
- Expand chemical space coverage to include under-represented compound classes [14]
- Develop standardized protocols for data curation and annotation [14]
3. Enhance Methodological Rigor
- Replace simplistic success rate calculations with nuanced risk assessment models [29]
- Develop standardized metrics that reflect real-world utility rather than theoretical performance [4]
- Implement improved statistical methodologies that account for program-specific factors [29]
4. Foster Cross-Community Collaboration
- Encourage collaboration between computational and experimental researchers [27]
- Establish partnerships between academia, industry, and regulatory agencies [4]
- Develop shared standards for data reporting and method documentation [28]
The implementation of this roadmap requires commitment across the research community but offers substantial rewards. As noted in recent literature, pairing "robust benchmarking with modern cheminformatic and bioinformatic tools like molecular dynamics simulations and machine learning" presents "a clear opportunity to raise the standard of computer-aided drug discovery" [4]. By addressing current gaps through sustained, community-wide effort, we can accelerate the translation of computational innovations into practical solutions for chemical and pharmaceutical challenges.
How to Benchmark: Frameworks, Tools, and Real-World Applications in Drug Discovery
Within computational chemistry and quantitative structure-activity relationship (QSAR) modeling, benchmarking serves as the critical process for objectively evaluating and validating computational models against known experimental data. This practice is fundamental for assessing model predictivity, reliability, and applicability domain, ensuring that in silico predictions can be trusted for decision-making in drug discovery and chemical safety assessment [1]. The impossibility of conducting experimental tests on all compounds due to cost and time constraints further underscores the necessity of robust computational methods [14]. A systematic benchmarking workflow, from rigorous data curation to comprehensive performance assessment, provides researchers, regulatory authorities, and industry professionals with a framework to identify optimal computational tools for predicting crucial physicochemical (PC) and toxicokinetic (TK) properties [14].
Foundational Principles of Model Validation
Validation of computational results against experimental data is paramount. This process ensures the accuracy and reliability of computational models, allowing researchers to confidently predict molecular properties and behaviors [1]. Key components of this validation include benchmarking, which evaluates models against known experimental results; model validation, which assesses how well computational predictions align with experimental observations; and error analysis, which quantifies discrepancies [1]. Without this rigorous process, models risk producing inaccurate predictions, leading to flawed scientific conclusions and poor decision-making in critical applications like drug development, where 40–60% of drug failures in clinical trials stem from PC and bioavailability deficiencies [14].
Error Analysis and Statistical Assessment
Comprehensive error analysis is essential for understanding model limitations. Systematic errors introduce consistent bias from improperly calibrated instruments or flawed theoretical assumptions, while random errors cause unpredictable fluctuations and can be reduced by increasing sample size [1]. Statistical techniques for validation include:
- Descriptive statistics (mean, median, standard deviation) to summarize dataset features
- Inferential statistics to draw conclusions about populations based on sample data
- Regression analysis to model relationships between variables
- Confidence intervals to provide plausible value ranges for population parameters [1]
Advanced approaches like machine learning techniques (random forests, neural networks) can identify complex patterns in large datasets, while Bayesian statistics incorporate prior knowledge and update probabilities as new data becomes available [1].
Stage 1: Data Curation and Standardization
High-quality molecular datasets are the foundation of reliable QSAR modeling and drug discovery [33]. The data curation process transforms raw, often inconsistent chemical data into standardized, ready-to-use datasets for cheminformatic analysis. This initial stage is critical, as many molecular databases contain inaccuracies such as invalid structures, duplicates, and experimental outliers that compromise model performance and reproducibility [33] [14]. Automated workflows help researchers retrieve chemical data (SMILES) from the web, check their correctness, and curate them to produce consistent datasets [34].
Data Collection and Curation Protocols
A robust curation procedure involves multiple steps to ensure data integrity. For substances lacking SMILES notation, isomeric SMILES should be retrieved using the PubChem PUG REST service from CAS numbers or chemical names [14]. The subsequent standardization and curation process should implement an automated procedure that addresses several key aspects:
Table 1: Key Steps in Molecular Data Curation
| Curation Step | Description | Implementation |
|---|---|---|
| Structure Validation | Identify and remove inorganic/organometallic compounds, mixtures, and compounds with unusual elements | RDKit Python package functions [14] |
| Salt Neutralization | Remove counterions to standardize to the parent structure | Automated in-house procedures [14] |
| Duplicate Removal | Identify and remove duplicates at SMILES level | Structural comparison algorithms [33] |
| Outlier Detection | Remove intra- and inter-outliers with inconsistent values | Z-score calculation (Z-score >3 considered outliers) [14] |
| Unit Standardization | Convert all data to consistent units for comparison | Appropriate conversion factors applied across datasets [14] |
For duplicate compounds, specific protocols must be followed. With continuous data, duplicates with a standardized standard deviation (standard deviation/mean) greater than 0.2 should be considered ambiguous and removed, while experimental values should be averaged if their difference is lower [14]. For binary classification data, only compounds with the same response values should be retained [14]. Tools like MEHC-curation implement a three-stage pipeline (validation, cleaning, normalization) with integrated duplicate removal and error tracking, making high-quality curation accessible to non-experts [33].
Data Curation Workflow
The following diagram illustrates the comprehensive data curation workflow from initial collection to finalized datasets:
Stage 2: Chemical Space Analysis and Applicability Domain
The applicability of benchmarking results is strictly limited to the chemical space covered by the datasets used for model evaluation [14]. Analyzing this chemical space ensures that validation results remain relevant to the specific categories of chemicals under investigation, such as pharmaceuticals, industrial chemicals, or natural products. Understanding the applicability domain (AD) of QSAR models is crucial for identifying when models can provide reliable predictions for query chemicals based on their similarity to the training set compounds [14].
Chemical Space Mapping Methodology
To obtain a meaningful view of the chemical space covered by validation datasets, chemicals should be plotted against a reference chemical space encompassing main categories of real-life interest. This reference space should include data from:
- The ECHA database of substances registered under REACH as representative of industrial chemicals
- The Drug Bank as representative of approved drugs
- The Natural Products Atlas as representative of natural chemical products [14]
The technical process for chemical space analysis involves:
- Standardization of all compound structures
- Fingerprint generation using functional connectivity circular fingerprints (FCFP) with a radius of 2 folded to 1024 bits computed using CDK
- Dimensionality reduction via principal component analysis (PCA) with two components applied to the descriptor matrix
- Visualization by plotting each collected dataset on the obtained two-dimensional chemical space defined by the PCA to determine coverage of relevant chemical categories [14]
This analysis confirms the validity of benchmarking results for specific chemical categories and helps researchers select appropriate models for their specific chemical classes of interest.
Stage 3: Model Training and Hyperparameter Tuning
With curated datasets and understood chemical space, the workflow proceeds to model development. Machine learning methods for developing classification QSAR models should incorporate calculation and selection of chemical descriptors, tuning of model hyperparameters, and methods to handle data unbalancing [34]. Automated workflows implementing six machine learning methods can efficiently develop QSAR models, with the additional capability to predict external chemicals [34].
Machine Learning Workflow for QSAR Modeling
The following diagram illustrates the comprehensive model training and validation process:
Stage 4: Performance Assessment and Benchmarking Metrics
The performance assessment stage quantitatively evaluates model predictivity using appropriate statistical metrics and validation procedures. For QSAR models predicting PC and TK properties, benchmarking typically emphasizes the performance of models inside their applicability domain [14]. This external validation provides the most realistic assessment of how models will perform on new, previously unseen chemicals.
Key Performance Metrics for Model Benchmarking
Different metrics are required for regression (continuous) and classification (categorical) models:
Table 2: Performance Metrics for Computational Model Assessment
| Model Type | Key Metrics | Interpretation and Application |
|---|---|---|
| Regression Models | R² (Coefficient of Determination) | Proportion of variance explained by the model; R² average of 0.717 for PC properties and 0.639 for TK properties reported in benchmarks [14] |
| Mean Absolute Error (MAE) | Average magnitude of errors between predicted and experimental values [1] | |
| Root Mean Square Error (RMSE) | Standard deviation of prediction errors, giving higher weight to large errors [1] | |
| Classification Models | Balanced Accuracy | Average accuracy between classes; average of 0.780 reported for TK properties [14] |
| Sensitivity and Specificity | Ability to correctly identify positive and negative cases, respectively | |
| Area Under ROC Curve (AUC-ROC) | Overall classification performance across all thresholds |
Performance analysis should also consider computational efficiency, with processing speed and scalability being important factors for practical application [33].
Implementation Framework: Tools and Reagents for Benchmarking
Successful implementation of a benchmarking workflow requires specific computational tools and resources. Prioritization should be given to freely available public software and tools that allow batch predictions for large datasets, evaluation of model applicability domain, and have publicly available training sets [14].
Essential Research Reagents and Computational Tools
Table 3: Essential Computational Tools for Benchmarking Workflows
| Tool/Resource | Function and Application | Key Features |
|---|---|---|
| KNIME Workflows | Automated data curation and ML model development [34] | Implements six machine learning methods; handles descriptor calculation, hyperparameter tuning, data unbalancing [34] |
| MEHC-Curation | Python framework for molecular dataset curation [33] | Three-stage pipeline (validation, cleaning, normalization); duplicate removal; error tracking [33] |
| OPERA | Open-source QSAR model battery [14] | Predicts various PC properties, environmental fate parameters, and toxicity endpoints; AD assessment using leverage and vicinity methods [14] |
| RDKit | Cheminformatics and machine learning software [14] | Chemical structure standardization; descriptor calculation; integration into Python workflows [14] |
| PubChem PUG | Chemical structure retrieval [14] | Access to chemical structures via CAS numbers or names; programmatic access via REST API [14] |
Reporting Standards and Best Practices
Transparent and systematic reporting of computational models facilitates their regulatory acceptance and use [35]. Standardized reporting formats help overcome barriers to broader model adoption, particularly for regulatory purposes. Adapted QSAR Model Reporting Formats (QMRF) provide consistent frameworks for describing models developed for nanomaterials and other chemical categories [35].
Best practices for benchmarking studies include:
- Documenting model applicability domain for all predictions
- Providing comprehensive performance metrics for both internal and external validation
- Reporting chemical space coverage to clarify model appropriateness for specific chemical categories
- Implementing model documentation in publicly accessible data catalogs to enhance reproducibility and transparency [35]
These standards ensure that benchmarking results are interpretable, comparable across studies, and suitable for informing regulatory decisions and scientific conclusions.
A systematic benchmarking workflow encompassing data curation, chemical space analysis, model training, and comprehensive performance assessment is indispensable for developing reliable computational chemistry models. By implementing the structured approach outlined in this guide—from initial data validation through final model reporting—researchers can establish robust, transparent, and reproducible modeling practices. This rigorous methodology enables the identification of optimal computational tools for predicting crucial chemical properties, ultimately accelerating drug discovery, chemical safety assessment, and regulatory decision-making while maintaining scientific rigor and transparency throughout the model development and validation lifecycle.
The rapid evolution of machine learning (ML) in computational chemistry has created an urgent need for standardized benchmarking frameworks. Machine-learned interatomic potentials (MLIPs) promise to revolutionize atomistic simulations by delivering quantum-level accuracy for large molecular systems at a fraction of the computational cost of traditional electronic structure methods [36] [7]. However, the field has historically lacked standardized and comprehensive tools for evaluating model performance, creating significant challenges in consistently discovering, comparing, and applying these models across diverse scenarios [37] [7]. This benchmarking gap hinders reproducibility, obscures genuine performance differences between models, and ultimately slows progress toward reliable, transferable potentials for complex molecular systems.
Standardized benchmarking addresses a fundamental mismatch between how MLIPs are validated and how they are ultimately used. While models are typically trained and evaluated using static error metrics on carefully curated quantum-mechanical datasets, real-world applications like molecular dynamics (MD) drive systems into regions of configuration space that are sparsely represented in training data [7]. Consequently, models with similar training accuracy often diverge significantly during long-timescale simulations or when predicting emergent physical behavior [7]. Benchmarking suites bridge this gap by providing rigorous, simulation-based evaluation protocols that reflect real-world scientific demands.
The Benchmarking Landscape: Frameworks for Computational Chemistry
The growing recognition of benchmarking importance has spurred development of several specialized frameworks, each designed to address distinct challenges within computational chemistry and materials science. The table below summarizes the prominent benchmarking frameworks available to researchers.
Table 1: Overview of Standardized Benchmarking Frameworks in Computational Chemistry
| Framework Name | Primary Focus | Key Features | Supported Systems |
|---|---|---|---|
| MLIPAudit [36] [37] [7] | General-purpose MLIP evaluation | Holistic metrics, leaderboard, modular design | Organic compounds, molecular liquids, proteins, peptides |
| Weighted Ensemble Benchmark [38] | Protein molecular dynamics methods | Enhanced sampling analysis, >19 evaluation metrics | Diverse proteins (10-224 residues) |
| CatBench [39] | Adsorption energy for catalysis | Multi-class anomaly detection, practical reliability focus | Heterogeneous catalysis, small to large molecules |
| ChemBench [26] | Chemical knowledge of LLMs | Automated evaluation, >2,700 question-answer pairs | Large language models for chemistry |
| MLIP Arena [7] | MLIP physical awareness and stability | Leaderboard, focuses on known failure modes | Materials and molecular systems |
MLIPAudit: A Comprehensive Framework for MLIP Evaluation
MLIPAudit represents a significant advance as an open, curated, and modular benchmarking suite designed specifically to assess MLIP accuracy across diverse application tasks [36] [37]. It shifts the evaluation focus from model-centric testing to systematic validation and comparison, addressing the critical need for standardized and reproducible evaluation protocols that go beyond basic error metrics [7].
The framework offers a diverse collection of benchmark systems, including small organic compounds, molecular liquids, proteins, and flexible peptides, along with pre-computed results for range of pre-trained models [36] [7]. Its modular architecture allows for easy expansion and community contribution, fostering an open-source approach that accelerates progress in MLIP development [36]. A continuously updated leaderboard on HuggingFace tracks performance across benchmarks, enabling direct comparison on downstream tasks and providing researchers with clear guidance for model selection [37] [7].
Specialized Benchmarking Frameworks
Other frameworks address more specialized needs within the computational chemistry ecosystem:
Weighted Ensemble Benchmarking Framework: This approach systematically evaluates protein MD methods using enhanced sampling analysis via WESTPA (Weighted Ensemble Simulation Toolkit with Parallelization and Analysis) [38]. It includes a dataset of nine diverse proteins ranging from 10 to 224 residues and computes over 19 different metrics and visualizations, including structural fidelity, slow-mode accuracy, and statistical consistency [38].
CatBench: This framework specializes in benchmarking MLIP performance for predicting adsorption energies in heterogeneous catalysis [39]. It employs multi-class anomaly detection to ensure rigorous benchmarking for practical deployment and has tested 13 ML models on over 47,000 reactions, with the best models achieving approximately 0.2 eV accuracy [39].
ChemBench: While not focused on MLIPs, this framework addresses the growing need to evaluate large language models (LLMs) in chemistry, providing an automated framework with over 2,700 question-answer pairs to assess chemical knowledge and reasoning against human expert performance [26].
Core Methodologies and Experimental Protocols
MLIPAudit's Evaluation Methodology
MLIPAudit employs a comprehensive evaluation strategy that moves beyond simple energy and force errors to reflect real-world simulation demands [36] [7]. The benchmark incorporates multiple assessment dimensions:
Energy and Force Accuracy: Fundamental validation against quantum-mechanical reference data using root-mean-square-error (RMSE) and mean-absolute-error (MAE) for energies and atomic forces on held-out validation datasets [7].
Model Stability and Transferability: Assessment of physical soundness under static extreme deformations and molecular dynamic stability under extreme temperatures and pressures [40].
Downstream Application Performance: Evaluation on tasks relevant to practical applications, including prediction of thermochemical properties of equilibrium structures and forces of far-from-equilibrium structures [40].
The benchmark workflow follows a systematic pipeline from system preparation through multiple assessment dimensions to quantitative divergence metrics, providing a holistic view of model capabilities and limitations.
Table 2: Key Experimental Metrics in Modern MLIP Benchmarks
| Metric Category | Specific Metrics | Purpose | Typical Methods |
|---|---|---|---|
| Static Accuracy | Energy MAE/RMSE, Force MAE/RMSE | Measure interpolation accuracy on QM data | Regression against DFT reference |
| Dynamic Stability | Simulation longevity, Energy conservation | Assess stability during MD simulations | Molecular dynamics simulations |
| Property Prediction | Radial distribution functions, Vibrational frequencies | Evaluate emergent physical behavior | Comparison to experimental or QM data |
| Sampling Efficiency | Conformational coverage, Rare event capture | Measure effectiveness for enhanced sampling | Weighted ensemble, Metadynamics |
| Robustness | Performance on out-of-domain systems | Test transferability and extrapolation | Cross-dataset validation |
Enhanced Sampling and Weighted Ensemble Methods
The weighted ensemble benchmarking framework employs sophisticated enhanced sampling methodologies to address timescale limitations in molecular simulations [38]. The protocol involves:
Progress Coordinate Definition: Using Time-lagged Independent Component Analysis (TICA) to derive progress coordinates that efficiently map conformational space [38].
Walker Propagation: Running multiple replicas of a system with periodic resampling based on user-defined metrics of conformational space coverage [38].
Comprehensive Analysis: Computing Wasserstein-1 and Kullback-Leibler divergences across multiple analyses, including TICA energy landscapes, contact map differences, and distributions for radius of gyration, bond lengths, angles, and dihedrals [38].
This approach enables fast and efficient exploration of protein conformational space, capturing critical transitions and rare events within tractable timeframes [38].
Successful implementation of standardized benchmarking requires familiarity with key software tools, datasets, and computational resources. The following toolkit provides researchers with essential components for effective MLIP evaluation.
Table 3: Essential Research Reagents and Resources for MLIP Benchmarking
| Resource Type | Examples | Function and Application | Access |
|---|---|---|---|
| Benchmarking Suites | MLIPAudit, CatBench, Weighted Ensemble Framework | Standardized evaluation and model comparison | GitHub, PyPI |
| Reference Datasets | Open Molecules 2025 (OMol25), MP-ALOE, MatPES | Training and validation data for MLIP development | Public repositories |
| Simulation Software | OpenMM, ASE (Atomic Simulation Environment) | Molecular dynamics engines and calculators | Open source |
| Enhanced Sampling Tools | WESTPA, PLUMED | Rare event sampling and free energy calculations | Open source |
| MLIP Architectures | MACE, ChgNet, ANI-1, CGSchNet | Pre-trained models and training frameworks | GitHub, model hubs |
| Leaderboards | MLIPAudit Leaderboard, MLIP Arena | Performance tracking and model comparison | HuggingFace, specialized portals |
Critical Datasets for Training and Validation
The quality of MLIP benchmarking depends significantly on the underlying datasets used for training and validation:
Open Molecules 2025 (OMol25): An unprecedented dataset of over 100 million 3D molecular snapshots calculated with density functional theory (DFT), featuring systems up to 350 atoms with broad chemical diversity across biomolecules, electrolytes, and metal complexes [6].
MP-ALOE: A dataset of nearly 1 million DFT calculations using the accurate r2SCAN meta-GGA functional, covering 89 elements and created using active learning to primarily consist of off-equilibrium structures [40].
MatPES: A public r2SCAN dataset for UMLIPs that samples structures from 300K molecular dynamics trajectories, providing a balance between near-equilibrium and off-equilibrium configurations [40].
These datasets enable comprehensive benchmarking across different chemical spaces and physical regimes, from equilibrium properties to far-from-equilibrium forces.
Visualizing Benchmarking Workflows
MLIPAudit Benchmarking Pipeline
Weighted Ensemble Sampling Methodology
Standardized benchmarking suites represent a critical infrastructure for advancing computational chemistry and drug discovery research. Frameworks like MLIPAudit, with their comprehensive, open, and modular design, are establishing much-needed reference points for evaluating MLIP performance across diverse molecular systems and application scenarios [36] [37] [7]. The integration of holistic metrics—spanning static accuracy, dynamic stability, property prediction, and transferability—provides researchers with a more complete picture of model capabilities and limitations than traditional error metrics alone.
The future of benchmarking in computational chemistry will likely involve continued expansion of benchmark systems to cover broader chemical spaces, increased emphasis on real-world application tasks, and tighter integration with experimental validation. As these frameworks evolve and gain wider adoption, they will accelerate progress toward more reliable, robust, and physically accurate machine learning potentials, ultimately enabling transformative advances in molecular modeling, materials design, and drug development.
Benchmarking Physicochemical and Toxicokinetic Property Predictors for ADMET
In computational chemistry and drug discovery, benchmarking is the systematic process of evaluating and comparing the performance of predictive models against standardized datasets and validation protocols. This practice is fundamental to model validation research, transforming theoretical algorithms into trusted tools for decision-making. For Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties, benchmarking provides critical assessment of model reliability in predicting physicochemical (PC) properties like solubility and lipophilicity, and toxicokinetic (TK) properties that define how the body handles chemicals over time. The ultimate goal is to establish confidence in computational predictions, thereby reducing reliance on costly and time-consuming experimental approaches, particularly those involving animal testing [14].
The process addresses key challenges in the ADMET prediction domain, including varying data quality, model applicability across different chemical spaces, and the need for standardized evaluation metrics. As the field evolves with more sophisticated machine learning (ML) and artificial intelligence (AI) approaches, rigorous benchmarking becomes increasingly vital for separating incremental improvements from genuine advances [41] [42]. This whitepaper examines current methodologies, performance landscapes, and best practices in benchmarking PC and TK property predictors to guide researchers, scientists, and drug development professionals in selecting and implementing these crucial tools.
Methodological Framework for Benchmarking Studies
Data Collection and Curation Protocols
The foundation of any robust benchmarking study lies in rigorous data collection and curation. Current best practices involve:
Multi-source Data Aggregation: Gathering experimental data from diverse public sources such as the Therapeutics Data Commons (TDC), ChEMBL, PubChem, and specialized databases like the Endocrine Disruptor Knowledgebase (EDKB) for specific endpoints [41] [43] [44]. The PharmaBench initiative, for instance, compiled 156,618 raw entries from 14,401 bioassays through an innovative data mining approach that incorporated a multi-agent system based on Large Language Models (LLMs) to identify experimental conditions [44].
Comprehensive Data Cleaning: Implementing standardized protocols to address common data issues including inconsistent SMILES representations, duplicate measurements with varying values, and ambiguous binary labels across train and test sets [41]. The cleaning workflow typically includes:
- Removal of inorganic salts and organometallic compounds
- Extraction of organic parent compounds from salt forms
- Tautomer standardization for consistent functional group representation
- SMILES canonicalization
- De-duplication with consistency checks (keeping first entry if target values are consistent, or removing entire groups if inconsistent) [41]
Chemical Space Analysis: Evaluating the representation of the validation datasets against reference chemical spaces covering major categories of interest (e.g., drugs from DrugBank, industrial chemicals from ECHA database, natural products from Natural Products Atlas) using techniques like principal component analysis (PCA) applied to molecular fingerprints [14].
Model Evaluation Metrics and Statistical Approaches
Comprehensive benchmarking employs multiple evaluation metrics to provide a complete picture of model performance:
For Regression Tasks (e.g., solubility, lipophilicity):
For Classification Tasks (e.g., Ames mutagenicity, hERG inhibition):
Advanced Statistical Validation:
- Integration of cross-validation with statistical hypothesis testing to add a layer of reliability to model assessments [41]
- Application of confidence intervals and Bayesian statistics to incorporate prior knowledge and update probabilities as new data becomes available [1]
- Use of ANOVA to compare means across multiple groups or models [1]
Applicability Domain Assessment
A crucial aspect of benchmarking is evaluating model performance within its applicability domain (AD) – the chemical space where the model makes reliable predictions. This involves:
- Leverage and Vicinity Methods: Identifying reliable predictions based on the distance of query chemicals from the training set [14]
- Domain of Applicability (DoA) Analysis: Ensuring the validation set represents a diverse set of molecular structures, chemical classes, and physicochemical properties [43]
- Uncertainty Quantification: More advanced tools provide confidence estimates for predictions, which is particularly important for decision-making in drug discovery pipelines [46] [47]
Current Landscape of ADMET Prediction Tools
Performance Comparison of Computational Tools
Recent comprehensive benchmarking studies have evaluated numerous software tools implementing QSAR models for PC and TK properties. The table below summarizes the performance of selected tools across key properties:
Table 1: Performance Comparison of ADMET Prediction Tools
| Property | Tool | Algorithm | Performance | Dataset Size |
|---|---|---|---|---|
| Caco-2 Permeability | Admetica [45] | Chemprop | MAE: 0.317, R²: 0.701 | 910 |
| PPBR | Admetica [45] | Chemprop | MAE: 6.919, R²: 0.609 | 2,790 |
| Lipophilicity | Admetica [45] | Chemprop | MAE: 0.399, R²: 0.748 | 4,200 |
| Solubility | Admetica [45] | Chemprop | MAE: 0.714, R²: 0.788 | 9,982 |
| hERG Inhibition | Admetica [45] | Chemprop | Balanced Accuracy: 0.854, ROC AUC: N/A | 22,249 |
| AMES Mutagenicity | PharmaBench [44] | Various | Classification metrics | 9,139 |
| CYP Inhibition | ADMET-AI [47] | Chemprop-RDKit | Best average rank on TDC Leaderboard | Varies by CYP |
Overall benchmarking results indicate that models for PC properties (average R² = 0.717) generally outperform those for TK properties (average R² = 0.639 for regression, average balanced accuracy = 0.780 for classification) [14]. This performance gap highlights the greater complexity of biological systems compared to physicochemical relationships.
Emerging Trends and Top Performers
The ADMET prediction landscape has evolved significantly with the integration of deep learning approaches:
ADMET-AI: Currently leads the TDC ADMET Leaderboard with the best average rank across 22 datasets. It employs a Chemprop-RDKit architecture that combines graph neural networks with 200 RDKit-computed molecular features, and provides contextualized predictions by comparing compounds to approved drugs from DrugBank [47].
Admetica: An open-source solution offering a comprehensive set of predictive models under the MIT license. It provides both command-line and web server interfaces and demonstrates competitive performance across multiple endpoints [45].
Commercial Platforms: Tools like ADMET Predictor remain widely used in industry, offering over 175 predicted properties with confidence estimations and applicability domain assessment [46] [48].
Recent approaches have emphasized multi-task learning (training single models on multiple ADMET properties) and ensembling (combining predictions from multiple models) to improve performance and robustness [47].
Experimental Protocols in Benchmarking Studies
Standardized Workflow for Model Validation
A robust benchmarking protocol follows a systematic sequence of experimental steps:
Table 2: Key Stages in Benchmarking Experimental Design
| Stage | Key Activities | Outputs |
|---|---|---|
| 1. Data Preparation | Data collection, cleaning, standardization, splitting | Curated datasets with train/validation/test splits |
| 2. Feature Selection | Evaluation of molecular representations (fingerprints, descriptors, embeddings) | Optimal feature sets for specific endpoints |
| 3. Model Training | Hyperparameter optimization, cross-validation, ensemble creation | Trained models with optimized architecture |
| 4. Statistical Evaluation | Performance metrics calculation, hypothesis testing, uncertainty quantification | Comprehensive model assessment report |
| 5. Practical Validation | External dataset testing, cross-dataset evaluation, real-world scenario assessment | Validation of model utility in practical applications |
The following diagram illustrates the complete experimental workflow for benchmarking ADMET predictors:
Data Curation Methodology
The data curation process is critical for reliable benchmarking. A comprehensive approach includes:
Structure Standardization: Using tools like the standardisation tool by Atkinson et al. (with modifications to include boron and silicon in organic elements definition) to create consistent SMILES representations [41].
Duplicate Handling:
- For continuous data: Removing duplicates with standardized standard deviation > 0.2, averaging if difference is lower
- For binary classification: Retaining only compounds with consistent response values [14]
Outlier Detection:
- Intra-outlier identification using Z-score method (removing points with Z-score > 3)
- Inter-outlier detection by comparing values across datasets for the same property [14]
This rigorous curation process typically results in the removal of 10-25% of initial compounds due to various quality issues [41] [14].
Advanced Validation: Cross-Dataset and Practical Scenario Testing
Beyond standard train-test splits, sophisticated benchmarking incorporates:
Scaffold Splitting: Separating compounds based on molecular scaffolds to assess performance on structurally novel compounds [41]
Cross-Dataset Validation: Training models on one data source and evaluating on a different source for the same property, which better mimics real-world application challenges [41]
Temporal Splitting: Evaluating models on data collected after the training data to simulate real-world deployment conditions [41]
These advanced validation techniques provide a more realistic assessment of model performance in practical drug discovery settings where compounds of interest often differ structurally from those in training data.
Table 3: Essential Research Reagents for ADMET Benchmarking Studies
| Resource Category | Specific Tools | Function | Key Features |
|---|---|---|---|
| Benchmarking Platforms | TDC ADMET Leaderboard [41] [47] | Standardized comparison of models across multiple ADMET endpoints | 22 curated datasets, standardized splits, leaderboard tracking |
| Open-source Packages | ADMET-AI [47], Admetica [45] | Ready-to-use models and frameworks for prediction | Pre-trained models, web interfaces, batch prediction capabilities |
| Data Resources | PharmaBench [44], TDC [41] [47] | Curated experimental data for training and validation | 52,482 curated entries across 11 ADMET datasets |
| Cheminformatics Tools | RDKit [41] [47], OPERA [14] | Molecular representation and descriptor calculation | Fingerprints, physicochemical descriptors, standardization utilities |
| Commercial Software | ADMET Predictor [46] [48] | Comprehensive property prediction with confidence estimation | Over 175 predicted properties, applicability domain assessment |
Validation Workflows and Decision Framework
The convergence of computational predictions and experimental validation is essential for establishing reliable ADMET models. The following diagram illustrates the integrated validation workflow:
This iterative process continues until models demonstrate adequate performance on external validation sets, with particular emphasis on performance within the model's applicability domain.
Benchmarking physicochemical and toxicokinetic property predictors for ADMET represents a cornerstone of computational chemistry validation research. The field has matured significantly from isolated model development to comprehensive, standardized evaluation frameworks. Current evidence suggests that while PC property predictions generally show higher accuracy than TK properties, ongoing advances in machine learning architectures—particularly graph neural networks augmented with molecular descriptors—are steadily closing this gap [47] [42].
The emergence of large-scale benchmarking initiatives like the TDC ADMET Leaderboard and PharmaBench, coupled with open-source tools such as ADMET-AI and Admetica, is democratizing access to state-of-the-art prediction capabilities while fostering transparency and reproducibility [47] [45] [44]. Future directions will likely focus on improving model interpretability, expanding coverage of novel chemical spaces, and enhancing uncertainty quantification—all critical for regulatory acceptance and more effective deployment in drug discovery pipelines.
For researchers and drug development professionals, selecting appropriate ADMET prediction tools should be guided by comprehensive benchmarking studies that evaluate performance across multiple endpoints, with particular attention to validation on external datasets and within relevant applicability domains. The ongoing integration of AI with traditional computational methods promises continued advancement in predicting these critical properties, ultimately accelerating the development of safer and more effective therapeutics.
Benchmarking represents a systematic process for evaluating computational model performance against standardized datasets and metrics, providing objective comparisons between different methodologies. In computational chemistry, particularly for Quantitative Structure-Activity Relationship (QSAR) models, rigorous benchmarking is indispensable for establishing model reliability for predicting chemical safety. With increasing regulatory reliance on New Approach Methodologies (NAMs) and growing ethical concerns regarding animal testing, robust validation frameworks ensure that QSAR predictions accurately identify hazards while minimizing false negatives that could endanger human health or ecosystems [14] [49].
This case study examines current benchmarking practices for QSAR models predicting environmental and human health safety endpoints. We analyze specific validation methodologies, performance metrics, and experimental protocols derived from recent large-scale benchmarking initiatives, providing researchers with practical guidance for implementing rigorous model assessment protocols aligned with regulatory requirements under frameworks like REACH and the European Chemicals Strategy for Sustainability [49].
Foundational Principles of QSAR Validation
Core Validation Components
Robust QSAR validation encompasses multiple interconnected components, each addressing distinct aspects of model reliability:
Applicability Domain (AD) Assessment: Determines the chemical space where the model can make reliable predictions based on the structural and physicochemical properties of its training data. Methods include leverage analysis and vicinity assessment to identify query chemicals outside the model's reliable prediction space [14].
External Validation: Evaluates model performance on completely independent datasets not used during model development, providing the most realistic assessment of predictive capability for new chemicals [14] [50].
Mechanistic Interpretability: Examines whether selected molecular descriptors align with established toxicological mechanisms, enhancing scientific confidence in predictions, particularly within Adverse Outcome Pathway (AOP) frameworks [49].
Performance Metrics for Different Tasks
Different QSAR applications require specialized performance metrics tailored to their specific contexts of use:
Table 1: Performance Metrics for QSAR Model Validation
| Application Context | Primary Metrics | Supplementary Metrics | Key Considerations |
|---|---|---|---|
| Regression Tasks (e.g., potency prediction) | R², RMSE, MAE | Q² (cross-validated R²) | Emphasis on prediction error magnitude and variance explanation [14] |
| Classification Tasks (e.g., toxicity hazard) | Balanced Accuracy, Sensitivity, Specificity | AUC, F₁-score | Traditional focus on balanced performance across classes [14] |
| Virtual Screening (hit identification) | Positive Predictive Value (PPV) | BEDROC, Early Enrichment | Prioritization of false positive minimization in top predictions [51] |
| Regulatory Acceptance | Sensitivity, Specificity | Applicability Domain Coverage | Emphasis on conservative hazard identification [52] |
For virtual screening of large chemical libraries, the traditional emphasis on balanced accuracy is shifting toward Positive Predictive Value (PPV), which better reflects the practical need to minimize false positives when only a small fraction of predicted actives can be experimentally tested [51].
Case Study: Large-Scale Benchmarking of Toxicokinetic and Physicochemical Property Predictors
Experimental Design and Workflow
A comprehensive benchmarking study evaluated twelve QSAR software tools for predicting 17 physicochemical (PC) and toxicokinetic (TK) properties relevant to chemical safety assessment. The methodology employed a rigorous multi-stage process to ensure unbiased evaluation [14]:
Table 2: Key Characteristics of the Benchmarking Study
| Aspect | Implementation |
|---|---|
| Software Tools Evaluated | 12 tools (prioritizing freely available options with batch prediction capability) |
| Properties Assessed | 17 PC and TK endpoints (e.g., logP, bioavailability, metabolic stability) |
| Validation Datasets | 41 curated external datasets (21 for PC, 20 for TK properties) |
| Chemical Space Coverage | 3 reference categories: REACH-registered substances, approved drugs, natural products |
| Data Curation | Standardization, salt removal, duplicate resolution, outlier detection (Z-score >3) |
| Statistical Analysis | Performance calculation restricted to chemicals within applicability domain |
The following workflow diagram illustrates the comprehensive benchmarking process:
Performance Outcomes and Key Findings
The benchmarking revealed significant differences in predictive performance across property types and software tools:
Table 3: Performance Summary of QSAR Tools for PC and TK Properties
| Property Category | Performance Level | Representative Tools | Notable Findings |
|---|---|---|---|
| Physicochemical Properties | R² average = 0.717 | OPERA, tools from project partners | Generally higher predictivity with more consistent performance across tools [14] |
| Toxicokinetic Properties (Regression) | R² average = 0.639 | Selected tools for specific endpoints | Moderate predictivity with higher endpoint-specific variability |
| Toxicokinetic Properties (Classification) | Balanced accuracy average = 0.780 | Tools with optimized AD assessment | Adequate classification performance with optimal tools identified per endpoint |
| Overall Assessment | Multiple optimal tools identified | Recurring best performers across properties | Tools with well-defined applicability domains generally more reliable |
Performance was notably higher for chemicals falling within models' applicability domains, highlighting the critical importance of AD assessment for reliable predictions. The study identified specific best-performing tools for each property endpoint, providing valuable guidance for researchers selecting computational tools for safety assessment [14].
Specialized Validation Protocols for Endpoint-Specific Models
Thyroid Hormone System Disruption
A comprehensive review of 86 QSAR models for thyroid hormone system disruption revealed distinct methodological patterns and research gaps. The analysis, covering literature from 2010-2024, identified molecular initiating events (MIEs) within the AOP framework as the primary modeling targets [49]:
Table 4: QSAR Modeling for Thyroid Hormone System Disruption
| Aspect | Current Status | Research Gaps |
|---|---|---|
| Modeling Targets | Inhibition of thyroperoxidase (TPO), binding to transthyretin (TTR) and thyroid receptors | Many MIEs in thyroid AOP poorly addressed or completely overlooked |
| Algorithm Preferences | Classification-based approaches dominant; simple algorithms prevalent | Limited use of complex machine learning methods despite their potential |
| Chemical Space Coverage | Focus on specific chemical classes (PCBs, PBDEs, bisphenols) | Limited coverage of diverse industrial chemicals and drug candidates |
| Mechanistic Interpretation | Some descriptor interpretation for TTR binding | Generally insufficient mechanistic alignment with biological pathways |
| Applicability Domain | Often inadequately defined | Need for explicitly defined ADs with broader chemical space coverage |
The validation approaches for thyroid disruption models typically employ chemical clustering and temporal validation to assess predictive performance for new chemical classes, with an emphasis on mechanistic interpretability of molecular descriptors [49].
Genotoxicity Assessment Using OECD QSAR Toolbox
A recent validation of OECD QSAR Toolbox profilers for genotoxicity assessment of pesticides demonstrated a specialized protocol for regulatory application:
Experimental Protocol:
- Dataset Curation: Compiled external validation dataset from MultiCASE Genotoxicity database, expanded with pesticide data from regulatory documents [52]
- Metabolism Simulation: Incorporated metabolism simulations using OECD QSAR Toolbox to assess influence on profiler performance
- Performance Assessment: Evaluated accuracy metrics for both AMES mutagenicity and in vivo micronucleus (MNT) endpoints
Key Findings:
- Absence of profiler alerts correlated well with experimentally negative outcomes (high specificity)
- Calculated accuracy varied considerably: 41%-78% for MNT-related profilers, 62%-88% for AMES-related profilers
- Incorporating metabolism simulations increased accuracy by 4-16% across endpoints
- Critical recommendation: Profilers alone should not be used directly for regulatory predictions; positive alerts require expert review and complementary evidence [52]
Table 5: Essential Research Reagent Solutions for QSAR Benchmarking
| Resource Category | Specific Tools | Function in Validation |
|---|---|---|
| Chemical Databases | PubChem, DrugBank, Natural Products Atlas, ChEMBL | Provide reference chemical spaces and experimental data for external validation [14] [50] |
| Descriptor Calculation | RDKit, DRAGON, PaDEL | Compute molecular descriptors and fingerprints for model development and AD assessment [14] [53] |
| Curated Benchmark Datasets | CARA benchmark, FS-Mol, OPERA models | Offer pre-validated datasets with specialized splitting schemes for realistic performance assessment [14] [50] |
| QSAR Platforms | OECD QSAR Toolbox, admetSAR, Way2Drug | Implement multiple validated models for specific endpoints with applicability domain assessment [14] [52] |
| Statistical Analysis | scikit-learn, KNIME, QSARINS | Provide algorithms for model building, feature selection, and performance metric calculation [53] |
Implementation Framework for Robust QSAR Validation
Standardized Workflow for Model Assessment
Based on the analysis of current benchmarking practices, we propose a comprehensive workflow for validating QSAR models for safety assessment:
Emerging Best Practices and Future Directions
Contemporary QSAR validation is evolving toward more sophisticated approaches:
Uncertainty Quantification: Implementation of conformal prediction frameworks that provide valid prediction intervals with specified confidence levels, addressing the need for reliable uncertainty estimates in regulatory decisions [54]
Multi-modal Model Integration: Development of unified frameworks like Uni-QSAR that combine 1D (SMILES), 2D (molecular graphs), and 3D (conformational) representations through ensemble and meta-learning, outperforming single-modality approaches [54]
Temporal and Chemical Drift Monitoring: Systematic assessment of model performance degradation over time as chemical libraries evolve, requiring regular model recalibration and monitoring of descriptor distribution shifts [54]
AI-Enhanced Validation: Integration of explainable AI (XAI) methods like SHAP and LIME to interpret model predictions and validate mechanistic plausibility, addressing the "black box" concern for complex machine learning models [53]
These advanced approaches collectively address the fundamental challenge in computational toxicology: establishing sufficient scientific confidence in QSAR predictions to support chemical safety decisions in the absence of experimental data.
Benchmarking serves as the cornerstone of progress in computational chemistry, providing the rigorous framework necessary to validate new methodologies and track field-wide advancements. In Structure-Based Drug Discovery (SBDD), the accuracy of predicting how a small molecule (ligand) binds to its protein target (pose prediction) and how tightly it binds (affinity prediction) directly impacts the efficiency and success rate of drug development [4]. The establishment of sustained, transparent benchmarking frameworks is not merely an academic exercise but a critical necessity for advancing computational drug design, mirroring the transformative role that the Critical Assessment of Structure Prediction (CASP) challenge played in revolutionizing protein structure prediction [4].
This whitepaper examines the current state, challenges, and future directions of benchmarking for pose and activity prediction (P-AP). We explore how robust benchmarking, when integrated with modern cheminformatic and bioinformatic tools like molecular dynamics simulations and machine learning, presents a clear opportunity to raise the standard of computer-aided drug discovery [4]. The discussion is framed within the broader context of computational chemistry model validation, emphasizing the need for community-driven efforts to overcome existing barriers and accelerate the development of therapeutic candidates.
The Critical Need for Benchmarking in SBDD
Despite its contributions to clinical candidate development, SBDD faces significant inconsistencies in reliably predicting ligand binding poses and affinities. A primary driver of this challenge is a pronounced shortage of high-quality experimental data required to develop and validate new computational methods [4]. For instance, one study highlighted that only 26% of noncovalently bound ligands and 46% of covalent inhibitors could be accurately regenerated within 2.0 Å RMSD of the experimental pose, underscoring the complexities of molecular simulation and docking approaches in real-world scenarios [4].
The field of small molecule drug discovery lacks a long-term, community-accepted benchmarking framework equivalent to CASP, which has continually driven progress in protein structure prediction for over three decades [4]. This absence makes it profoundly difficult for researchers to compare methods and track genuine improvements in key areas such as binding mode prediction and molecular dynamics accuracy. Without standardized evaluations, innovative computational techniques struggle to gain widespread adoption, as their purported advantages remain difficult to quantify objectively [4].
Table 1: Key Challenges in Benchmarking for Structure-Based Drug Discovery
| Challenge Category | Specific Issue | Impact on Model Validation |
|---|---|---|
| Data Quality & Availability | Shortage of high-quality experimental structures and affinity data | Limits development and validation of new computational methods [4] |
| Dataset Construction | Overlap between training and evaluation datasets | Inflates performance metrics, leading to overestimation of generalization [55] |
| Use of unconfirmed decoys and presence of non-physical poses | Introduces biases and artifacts that compromise assessment [4] | |
| System Complexity | Structurally complex or flexible binding sites | Challenges the predictive power of both physics-based and ML models [4] |
| Experimental Variability | Variability in experimental data quality (e.g., affinity measurements) | Creates unreliable ground truth labels for model training and testing [4] |
Current Methodologies and Limitations
Computational methods for predicting protein-ligand interactions generally involve two core stages: sampling (exploring possible ligand conformations and orientations within the binding site) and scoring (ranking these poses based on estimated binding affinity) [4]. These approaches have evolved from foundational techniques based on physical energy functions, knowledge-based potentials, and empirical terms to more recent strategies integrating machine learning (ML), deep learning, and even large language models [4] [56].
AI-driven methodologies are significantly enhancing key aspects of SBDD. Ligand binding site prediction has been refined using geometric deep learning and sequence-based embeddings [56]. For binding pose prediction, both sampling-based and regression-based deep learning models have evolved, alongside innovative protein-ligand co-generation frameworks as demonstrated by AlphaFold3 and related approaches [57] [56]. In scoring function development, AI-powered models now integrate physical constraints with deep learning to improve binding affinity estimation, leading to more robust virtual screening strategies [56].
Critical Limitations and Data Leakage
A fundamental issue plaguing the field is the overestimation of model performance due to shortcomings in benchmark design. The most critical problem is train-test data leakage, where the data used to evaluate a model is not sufficiently independent from the data used to train it. This severely inflates performance metrics and creates an unrealistic picture of a model's generalization capabilities [55].
This problem is pronounced in affinity prediction. Many deep-learning-based scoring functions are trained on the PDBbind database and evaluated using the Comparative Assessment of Scoring Function (CASF) benchmark. A 2025 study revealed a substantial level of data leakage between these datasets; nearly 600 high-similarity pairs were identified between PDBbind training and CASF complexes, affecting 49% of all CASF test complexes [55]. These similarities enable models to make accurate predictions through memorization of structural patterns rather than genuine understanding of protein-ligand interactions. Alarmingly, some models maintain comparable performance on CASF benchmarks even after omitting all protein or ligand information from their input, confirming that their predictions are not based on learning the underlying interactions [55].
Similar data leakage concerns exist for pose prediction. The good reported performance of many deep learning and cofolding methods can be partially attributed to similarities between training and test sets, particularly when using time splits on the PDBBind set where test sets contain identical or very similar proteins and ligands [57].
Table 2: Impact of Data Cleaning on Model Performance (CASF Benchmark)
| Model / Training Condition | Reported Performance (RMSE) | Performance on CleanSplit (RMSE) | Notes |
|---|---|---|---|
| GenScore (trained on original PDBbind) | Excellent | Marked drop | Performance drop confirms previous scores were driven by data leakage [55] |
| Pafnucy (trained on original PDBbind) | Excellent | Marked drop | Performance drop confirms previous scores were driven by data leakage [55] |
| GEMS (trained on PDBbind CleanSplit) | N/A | State-of-the-art | Maintains high performance on a genuinely independent test set, demonstrating true generalization [55] |
| Simple Search Algorithm (on uncleaned data) | Competitive with some DL models (Pearson R=0.716) | N/A | Finds most similar training complexes and averages their affinity, highlighting the role of memorization [55] |
Best Practices and Experimental Protocols for Robust Benchmarking
Designing Rigorous Benchmarking Experiments
A robust benchmarking protocol in SBDD must prioritize the independence of test data and the transparency of methodology. Key steps include:
- Strict Dataset Splitting: Implement structure-based clustering algorithms to create training and test sets that are strictly separated. This involves assessing protein similarity (using metrics like TM-score), ligand similarity (Tanimoto score), and binding conformation similarity (pocket-aligned ligand RMSD) to eliminate complexes with high similarity across splits [55]. The PDBbind CleanSplit protocol is an exemplar, which removes not only training complexes closely resembling test complexes but also those with highly similar ligands (Tanimoto > 0.9) to prevent ligand-based memorization [55].
- Blinded Evaluation: To ensure objectivity, evaluation should be performed on sequestered test sets where the true values (e.g., experimental affinities or poses) are hidden from the model developers until after predictions are submitted [4]. This approach mirrors the successful CASP challenge model.
- Comprehensive Data Curation: Prior to benchmarking, experimental data must be rigorously curated. This includes standardizing chemical structures (e.g., using RDKit), neutralizing salts, removing duplicates, and identifying and reconciling experimental outliers and ambiguous values across different data sources [14]. For affinity data, this may involve calculating Z-scores to flag and remove intra-dataset outliers and comparing values for the same compound across multiple datasets to remove inter-outliers [14].
- Application of the Applicability Domain (AD): The performance of a model should be interpreted in the context of its applicability domain—the chemical and structural space for which it was trained. Predictions for compounds outside the AD are less reliable. Benchmarking reports should distinguish between performance on the entire test set and performance only on those compounds falling within the models' AD [14].
Workflow for a Community Benchmarking Challenge
The following diagram illustrates a generalized workflow for organizing a community-wide benchmarking challenge, such as those needed for pose and affinity prediction, incorporating best practices for minimizing data leakage and ensuring fair model comparison.
Table 3: Key Resources for Benchmarking in SBDD
| Resource / Tool | Type | Primary Function in Benchmarking |
|---|---|---|
| PDBbind [55] | Database | Comprehensive collection of protein-ligand complexes with binding affinity data; used for training and testing scoring functions. |
| CASF Benchmark [55] | Benchmark Set | Standardized benchmark for the comparative assessment of scoring functions (affinity prediction) and docking (pose prediction). |
| PoseBusters [57] | Benchmark Tool | Used to validate the physical realism and chemical correctness of predicted protein-ligand poses. |
| RDKit [57] [14] | Cheminformatics Library | Open-source toolkit for cheminformatics, used for standardizing chemical structures, descriptor calculation, and molecular operations. |
| OPERA [14] | QSAR Tool | Open-source battery of QSAR models for predicting physicochemical properties and toxicity endpoints; includes applicability domain assessment. |
| AlphaFold3, Boltz, RFdiffusion [55] [57] | AI Prediction Tool | Cofolding and structure prediction tools used for generating protein-ligand complex structures and assessing pose prediction methods. |
Future Directions and Community Initiatives
The path forward for benchmarking in SBDD requires a concerted, community-wide effort. Key recommendations from leading researchers include:
- Introducing Blinded Evaluation Methods: Adopting blinded challenges, similar to CASP or the ASAP-Polaris-OpenADMET antiviral competition, for greater objectivity in assessing new methodologies [4] [57].
- Developing Diverse and Challenging Datasets: Moving beyond current benchmarks to create datasets that reflect real-world therapeutic targets, including membrane proteins, and challenging cases like activity cliffs (where small structural changes cause large affinity changes) [4].
- Encouraging Continuous Updates: Establishing frameworks for the continuous release of new benchmarking sets to keep pace with new experimental data and emerging target classes [4].
- Promoting Cross-Sector Collaboration: Fostering collaboration across academia, industry, and even competing commercial organizations to establish universally accepted standards and benchmarks [4].
- Integrating Cutting-Edge Technologies: Leveraging advanced molecular dynamics simulations, AI-based prediction tools, and large language models to create more accurate and efficient benchmarking pipelines [4] [56].
Addressing the generalization challenge remains paramount. Future work must focus on incorporating protein flexibility, allostery, and the ability to learn from diverse data types to build models that are robust across the vast and uncharted regions of chemical and biological space [56]. The ultimate goal is to develop benchmarking frameworks that not only measure performance on historical data but also reliably predict a method's success in prospective, real-world drug discovery projects.
Benchmarking in structure-based drug discovery is not a peripheral activity but a central driver of methodological innovation and reliability. The current state of the field, while advanced, is hampered by data leakage, dataset redundancy, and a lack of sustained community-wide benchmarking challenges. Addressing these issues through strict dataset splitting, blinded evaluations, and rigorous data curation—as exemplified by initiatives like PDBbind CleanSplit—is essential for obtaining an honest assessment of model performance and generalization.
The collective call to action from researchers across leading institutions is clear: the future of computational drug discovery depends on the establishment of transparent, continuous, and challenging benchmarking frameworks. By pairing these robust validation practices with modern AI-driven methodologies, the community can raise the standards of computer-aided drug design, ultimately accelerating the discovery of new therapeutic agents. For researchers in computational chemistry, medicinal chemistry, and SBDD, actively participating in and adhering to these benchmarking standards is crucial for strengthening the foundational tools of the trade.
Beyond Basic Metrics: Troubleshooting Poor Performance and Optimizing Benchmarking Protocols
Benchmarking serves as the cornerstone of progress in computational chemistry, enabling researchers to validate new methodologies, compare algorithmic performance, and establish trust in predictive models. Within the broader context of computational chemistry model validation research, benchmarking provides the essential framework for assessing whether new methods represent genuine advancements or merely exploit hidden biases in evaluation datasets. The reliability of computational models directly impacts critical applications across chemical sciences, from drug discovery and materials design to environmental risk assessment. As the field increasingly adopts data-driven approaches, rigorous benchmarking practices become paramount to distinguish between methodological improvements and statistical artifacts that fail to generalize beyond curated test sets.
The fundamental goal of benchmarking in computational chemistry is to provide a fair, reproducible, and scientifically meaningful evaluation of a model's predictive capability for its intended application domain. However, this process is fraught with challenges that can compromise validation outcomes. This technical guide examines three pervasive pitfalls—data leakage, overfitting, and inadequate applicability domain characterization—that routinely undermine benchmarking studies, drawing upon recent case studies and methodological analyses to illustrate their consequences and present mitigation strategies.
Data Leakage: The Silent Validity Killer
Data leakage occurs when information from outside the training dataset inadvertently influences the model development process, creating an over-optimistic assessment of performance that fails to generalize to truly novel data. This pitfall represents a fundamental breach of the core principle in machine learning: that models should be evaluated exclusively on data they have not encountered during training.
Case Study: Data Integrity Failures in LIT-PCBA
A recent audit of the widely-used LIT-PCBA virtual screening benchmark revealed severe data leakage that fundamentally compromises its utility for evaluating model performance [58]. The analysis identified multiple critical integrity failures:
- Cross-set Duplication: The benchmark contained 2,491 inactive compounds duplicated between training and validation sets, allowing models to "memorize" supposedly unseen data [58]
- Query Set Leakage: Three ligands in the query set—intended to represent unseen test cases—appeared identically in either training or validation splits [58]
- Structural Redundancy: For some targets, over 80% of query ligands were near duplicates (Tanimoto similarity ≥0.9) of training compounds, with ALDH1 alone containing 323 highly similar active pairs between training and validation sets [58]
The consequences of these leaks were profound. Researchers demonstrated that a trivial memorization-based model with no chemical intelligence could outperform sophisticated deep learning architectures like CHEESE simply by exploiting these benchmark artifacts [58]. This case illustrates how data leakage can invalidate an entire benchmark, calling into question previously reported state-of-the-art results.
Experimental Protocols to Prevent Data Leakage
Table 1: Methodological Safeguards Against Data Leakage
| Protocol | Implementation | Validation Technique |
|---|---|---|
| Temporal Splitting | Order data by publication date; train on earlier data, test on newer data | Assess performance degradation on chronological splits |
| Structural Similarity Analysis | Calculate Tanimoto coefficients between all training and test compounds | Remove test compounds with similarity > threshold (e.g., 0.85) to any training compound |
| Identity Checking | Exact duplicate detection using canonical SMILES and stereochemistry awareness | Comprehensive cross-set comparison with standardized representations |
| Domain-Informed Splitting | Group compounds by scaffold, protein target, or experimental assay before splitting | Ensure representative but non-overlapping splits across meaningful biological/chemical groupings |
Implementing these protocols requires specialized tools and careful experimental design. The structural similarity analysis should employ circular fingerprints (such as ECFP4 or FCFP4) with Tanimoto similarity calculations, systematically checking all train-test pairs [14]. Identity checking must account for stereochemistry and tautomeric forms by using standardized molecular representations (e.g., canonical isomeric SMILES) [50]. For benchmarks incorporating multiple data sources, provenance tracking becomes essential to avoid cross-contamination between supposedly independent datasets.
Overfitting: When Models Learn the Noise
Overfitting occurs when models learn dataset-specific artifacts rather than underlying chemical principles, resulting in impressive benchmark performance that masks poor generalization capability. This pitfall is particularly prevalent in computational chemistry due to the high-dimensional nature of chemical data and frequently limited dataset sizes.
The Dual Layer of Overfitting
In computational chemistry benchmarking, overfitting manifests at two distinct levels:
- Model-Level Overfitting: Traditional overfitting where a model with excessive complexity memorizes training examples rather than learning generalizable relationships
- Benchmark-Level Overfitting: Repeated tuning and evaluation on fixed benchmark datasets leads to indirect overfitting, where methodologies become increasingly specialized to particular benchmarks without genuine algorithmic improvements
The latter form is particularly insidious, as noted in analyses of materials informatics benchmarks, where performance rankings can become biased toward the data distribution of single sources like the Materials Project [59]. This creates a self-reinforcing cycle where methods appear to improve while actual predictive capability stagnates.
Quantitative Diagnostics and Mitigation Strategies
Table 2: Overfitting Detection and Prevention Metrics
| Metric | Application | Interpretation |
|---|---|---|
| Train-Test Performance Gap | Compare performance on training vs. test sets | Large gaps (>15-20%) indicate potential overfitting |
| Learning Curves | Monitor performance as training data increases | Plateauing test performance suggests insufficient data or model limitations |
| Cross-Validation Variance | Assess performance variation across folds | High variance indicates sensitivity to specific data partitions |
| Benchmark Diversity Score | Evaluate chemical space coverage of benchmarks | Low diversity increases overfitting risk; requires structural and property-based assessment |
Recent work on uncertainty quantification provides additional safeguards against overfitting. Techniques such as distance-to-training-set measures (e.g., Tanimoto similarity to nearest training compound) and predictive confidence intervals help identify extrapolations beyond reliable prediction regions [60]. For the CARA benchmark in compound activity prediction, researchers implemented specialized train-test splitting schemes that separate structurally similar compounds to better simulate real-world generalization [50].
Inadequate Applicability Domains: The Generalization Fallacy
The applicability domain (AD) of a model defines the chemical space within which its predictions are reliable. Inadequate characterization of AD represents a critical pitfall in benchmarking, as it leads to overestimation of model utility for real-world applications where chemical diversity exceeds benchmark coverage.
Case Studies in Applicability Domain Failures
Multiple recent studies highlight the consequences of inadequate AD characterization:
- In benchmarking studies on iminodiacetic acid (IDA), computational approaches completely failed to predict vibrational spectra at high frequencies (>2200 cm⁻¹) despite strong performance at lower frequencies, revealing critical method-specific limitations [61]
- For toxicity prediction models, performance dropped significantly when evaluating chemicals outside the training domain, particularly for industrial compounds versus pharmaceuticals [14]
- The CARA benchmark for compound activity prediction identified that biased protein exposure in training data—where some protein targets are vastly overrepresented—creates hidden generalization gaps [50]
These cases illustrate how models with apparently strong benchmark performance can fail catastrophically when applied to even slightly different chemical domains, highlighting the necessity of comprehensive AD assessment.
Experimental Protocols for Applicability Domain Characterization
Table 3: Applicability Domain Assessment Techniques
| Technique | Description | Implementation |
|---|---|---|
| Chemical Space Visualization | Project compounds into 2D space using dimensionality reduction | Principal Component Analysis (PCA) of molecular descriptors or fingerprints [14] |
| Distance-Based Measures | Quantify similarity to training set | Tanimoto similarity, Euclidean distance in descriptor space [60] |
| Domain-Specific Validation | Test performance on chemically distinct subsets | Stratified evaluation by molecular weight, scaffold, or functional groups [50] |
| Consensus Methods | Compare predictions across multiple algorithms | Flag compounds with high prediction variance as outside AD [60] |
The experimental workflow for comprehensive AD assessment begins with chemical space analysis using descriptors such as functional connectivity circular fingerprints (FCFP) followed by PCA to visualize coverage relative to relevant reference chemical spaces (e.g., drug databases, industrial chemicals, natural products) [14]. Performance should then be evaluated across stratified subsets of the chemical space, with particular attention to regions with sparse training data. For the ONTOX project, this approach enabled identification of robust QSAR models for toxicokinetic properties by explicitly evaluating performance within well-defined applicability domains [14].
Integrated Workflow for Robust Benchmarking
The following diagram illustrates a comprehensive benchmarking workflow that systematically addresses the three pitfalls discussed in this guide:
Integrated Benchmarking Workflow: This comprehensive workflow integrates safeguards against all three major pitfalls through sequential checking phases and parallel validation strategies.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 4: Essential Computational Tools for Robust Benchmarking
| Tool Category | Specific Examples | Function in Benchmarking |
|---|---|---|
| Cheminformatics Libraries | RDKit, CDK, OpenBabel | Molecular standardization, descriptor calculation, fingerprint generation [14] [50] |
| Similarity Metrics | Tanimoto coefficient, Euclidean distance | Quantifying molecular similarity for leakage checks and AD definition [60] [58] |
| Data Curation Tools | Custom Python scripts, PubChem PUG REST | Structure standardization, duplicate removal, outlier detection [14] |
| Visualization Frameworks | PCA, t-SNE, chemical space plots | Mapping dataset coverage and identifying gaps in chemical space [50] [14] |
| Uncertainty Quantification | Conformal prediction, ensemble variance | Assessing prediction reliability and defining applicability boundaries [60] |
The accelerating integration of computational methods across chemical research domains necessitates increasingly rigorous benchmarking practices. As demonstrated through multiple case studies, failures to address data leakage, overfitting, and applicability domain limitations can produce profoundly misleading conclusions about methodological capabilities. The experimental protocols and diagnostic frameworks presented in this guide provide a foundation for more robust validation approaches that better simulate real-world performance.
Future benchmarking efforts must prioritize comprehensive chemical space coverage, rigorous separation between model development and evaluation data, and transparent reporting of limitations. Initiatives like JARVIS-Leaderboard, which integrates multiple categories of methods and data modalities, represent promising directions for the field [59]. Similarly, the development of specialized benchmarks like CARA for compound activity prediction demonstrates the value of task-specific evaluation frameworks that mirror real-world application scenarios [50].
By adopting the integrated workflow and methodological safeguards outlined in this technical guide, researchers in computational chemistry and drug development can produce more meaningful, reproducible, and scientifically valid benchmark results that genuinely advance the field rather than merely optimizing for misleading metrics.
In the context of benchmarking computational chemistry models, error analysis is not merely a procedural step; it is the cornerstone of validation and reliability. For researchers and drug development professionals, understanding and distinguishing between systematic and random errors is critical for assessing the true performance of computational methods, such as Neural Network Potentials (NNPs) or density functional theory (DFT). This guide provides an in-depth technical framework for identifying, quantifying, and mitigating these errors, ensuring robust model validation within computational chemistry research.
Theoretical Foundations: Systematic vs. Random Error
In scientific research, measurement error is the difference between an observed value and the true value. These errors are broadly categorized into two types: random and systematic [62].
- Random Error: A random error is a chance difference between the observed and true values. It introduces unpredictable variability between different measurements of the same thing and is often considered "noise" that blurs the true "signal" [62]. Random error primarily affects the precision of a measurement, which refers to how reproducible the same measurement is under equivalent circumstances [62].
- Systematic Error: A systematic error is a consistent or proportional difference between the observed and true values. It skews measurements in a specific, predictable direction every time a measurement is made [63] [62]. Systematic error primarily affects the accuracy of a measurement, or how close the observed value is to the true value [62].
In research, systematic errors are generally considered a more significant problem than random errors. While random errors often cancel each other out when averaged over a large sample, systematic errors will consistently bias data away from the true value, potentially leading to false conclusions about the relationships between variables [62].
A Visual Analogy: The Dartboard
The concepts of accuracy and precision are effectively illustrated using a dartboard analogy [62]:
- High Accuracy, Low Precision: Darts are scattered around the bullseye (true value). Measurements are correct on average, but individual observations are variable.
- Low Accuracy, High Precision: Darts are clustered tightly together, but away from the bullseye. Measurements are reproducible but consistently biased.
- High Accuracy, High Precision: Darts are clustered tightly on the bullseye. Measurements are both correct and reproducible.
Understanding the origins of errors is the first step in mitigating them.
Random errors arise from unknown or unpredictable fluctuations [63] [62]. In computational chemistry, these can include:
- Natural Variations: Slight, inherent numerical instabilities in iterative algorithms like self-consistent field (SCF) convergence.
- Imprecise Instruments: Limitations in the convergence criteria or numerical integration grids used in quantum chemistry software.
- Poorly Controlled Procedures: Variations in initial geometry guess or conformational sampling that lead to different optimized structures and, consequently, energies.
Systematic errors skew results in a consistent direction [62]. Two quantifiable types are:
- Offset Error (Additive/Zero-Setting Error): Occurs when a scale isn't calibrated to a correct zero point, shifting all observed values by a fixed amount [62].
- Scale Factor Error (Multiplicative Error): Occurs when measurements consistently differ from the true value proportionally (e.g., by 10%) [62].
In computational chemistry, common sources include:
- Methodological Bias: The inherent limitations of a theoretical method. For example, a specific density functional (e.g., B97-3c) may systematically overestimate reduction potentials for organometallic species [8].
- Instrument Bias: A miscalibrated or consistently faulty piece of equipment used to collect experimental reference data.
- Sampling Bias: When a dataset overrepresents certain types of molecules (e.g., main-group organics) and underrepresents others (e.g., organometallics), leading to a model that performs poorly on the underrepresented class [8].
- Procedural Bias: Consistently incorrect usage of software, such as applying an implicit solvation model unsuitable for a specific solvent.
Methodologies for Error Quantification and Analysis
Robust error analysis relies on quantitative metrics and structured methodologies.
Core Quantitative Metrics for Error Analysis
The following metrics are essential for quantifying model performance and characterizing errors in benchmarking studies [8].
Table 1: Key Quantitative Metrics for Computational Model Benchmarking
| Metric | Formula | Interpretation | Relevance to Error Type | ||
|---|---|---|---|---|---|
| Mean Absolute Error (MAE) | Average magnitude of errors, directly interpretable in the target variable's units. | A high MAE indicates large overall error; consistent deviation suggests systematic error. | |||
| Root Mean Squared Error (RMSE) | Average magnitude of errors, but penalizes larger errors more heavily than MAE. | A high RMSE relative to MAE suggests the presence of large, unpredictable errors (high random error or outliers). | |||
| Coefficient of Determination (R²) | Proportion of variance in the observed data that is predictable from the model. | A low R² indicates poor model fit, often resulting from systematic bias or high random error (noise). |
Experimental Protocol: A Case Study in Reduction Potential Prediction
A detailed methodology from a benchmark study on predicting reduction potentials illustrates a robust error analysis workflow [8].
- Data Curation: Obtain experimental reduction-potential data for a diverse set of species (e.g., 192 main-group and 120 organometallic molecules) from a curated source [8].
- Computational Modeling:
- Geometry Optimization: Optimize the non-reduced and reduced structures of each species using the model(s) under investigation (e.g., NNPs, DFT functionals) [8].
- Solvent Correction: Input optimized structures into an implicit solvation model (e.g., CPCM-X) to obtain solvent-corrected electronic energies [8].
- Property Calculation: Calculate the predicted reduction potential as the difference in electronic energy (in eV) between the non-reduced and reduced structures.
- Error Quantification: Compare predicted values against experimental data using the metrics in Table 1 (e.g., MAE, RMSE, R²) [8].
- Comparative Analysis: Benchmark the performance of new models (e.g., OMol25 NNPs) against established low-cost methods (e.g., DFT functional B97-3c, semiempirical method GFN2-xTB) [8].
- Error Decomposition: Analyze performance disaggregated by chemical class (e.g., main-group vs. organometallic) to identify systematic biases [8].
A Framework for Error Mitigation
Different error types require different mitigation strategies.
Mitigating Random Error
- Take Repeated Measurements: In computational terms, this can involve running calculations with different initial conditions or random seeds and averaging results to converge on a more stable value [62].
- Increase Sample Size: Using larger, more comprehensive datasets for training and benchmarking reduces the impact of random variability and improves the statistical power of model evaluations [62].
- Control Variables: Carefully standardize computational protocols (e.g., convergence criteria, grid size, basis set) across all calculations to minimize unpredictable fluctuations [62].
Mitigating Systematic Error
- Triangulation: Use multiple independent methods or data sources to measure the same property. For example, corroborate NNP predictions with results from different DFT functionals or experimental data [62].
- Regular Calibration: Compare model predictions or instrument readings against known, standard reference values. For NNPs, this involves continuous benchmarking on high-quality, gold-standard datasets [62].
- Randomization: When constructing training and test sets, use random sampling to ensure they are representative of the broader chemical space, thereby reducing sampling bias [62].
- Blinding (Masking): Where possible, hide the condition assignment (e.g., the identity of the method being tested) during the data analysis phase to prevent experimenter expectancies from influencing the results [62].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for Error Analysis in Model Validation
| Tool / Reagent | Function in Error Analysis |
|---|---|
| Neural Network Potentials (NNPs) | Machine-learning models trained on quantum chemical data for fast, accurate energy and property predictions. Their performance must be benchmarked for systematic biases [8]. |
| Density Functional Theory (DFT) | A computational quantum mechanical method used to investigate electronic structure. Different functionals have known systematic errors [8]. |
| Semiempirical Quantum Mechanical (SQM) Methods | Approximate quantum mechanical methods parameterized from experimental data. Useful for rapid screening but can contain significant systematic biases [8]. |
| Implicit Solvation Models (e.g., CPCM-X) | Account for solvent effects in computations. The choice of model can be a source of systematic error in predicting solution-phase properties like reduction potential [8]. |
| Geometry Optimization Algorithms | Algorithms that locate stable molecular conformations. The choice of algorithm and convergence criteria can influence results and introduce random errors [8]. |
Workflow and Error Classification Diagrams
Computational Model Validation Workflow
The following diagram outlines a standard workflow for validating computational chemistry models, incorporating key steps for error analysis.
Model Validation and Error Analysis Workflow
Error Classification and Impact
This diagram classifies errors and illustrates their distinct impacts on data.
Classification of Measurement Errors
Within the rigorous framework of computational chemistry model validation, meticulous error analysis is paramount. Systematically differentiating between random and systematic errors enables researchers to move beyond simple performance metrics and understand the fundamental limitations and biases of their models. By applying the quantitative methods, detailed protocols, and targeted mitigation strategies outlined in this guide, scientists and drug developers can enhance the reliability of their computational predictions, thereby accelerating and de-risking the drug discovery process.
In computational chemistry and drug development, the relentless emergence of new methods and software packages presents researchers with a critical challenge: selecting the optimal tool for a given scientific problem. Benchmarking analysis, the process of rigorously comparing performance metrics to those of peers or competitors, is the primary strategy employed to address this challenge, aiming to identify best practices and opportunities for growth [64]. In an ideal scenario, a simple speed test would provide a clear, objective ranking of computational methods. However, the pursuit of such a straightforward comparison is fraught with complexity. Performance is multidimensional, and a myopic focus on raw speed can be not only misleading but scientifically detrimental. This whitepaper, framed within the broader context of model validation research, argues that effective benchmarking in computational chemistry must transcend simplistic speed comparisons. It must instead embrace a holistic, rigorously designed framework that evaluates a spectrum of performance criteria—including accuracy, robustness, scalability, and usability—against well-defined scientific objectives. The limitations of speed tests are not merely theoretical; they stem from intrinsic methodological and practical challenges that, if unaddressed, can compromise the validity of scientific conclusions and hinder progress in fields like drug discovery.
The Inherent Complexities of Computational Benchmarking
The Multifaceted Nature of "Performance"
Reducing performance to a single metric, such as computation time, provides an incomplete and potentially distorted view. A tool that is fastest but fails to converge for challenging molecular systems, or one that produces less accurate results, is of little value in rigorous research. True performance is a composite of several factors:
- Accuracy and Predictive Performance: This is the cornerstone of scientific utility. For a model predicting toxicokinetic properties, performance is measured by metrics like the coefficient of determination (R²) for regression or balanced accuracy for classification [14].
- Computational Efficiency: While this includes raw speed (e.g., time to a single-point energy calculation), it also encompasses scalability—how well the method performs as the problem size (number of atoms, electrons) or computational resources (core count) increases [65].
- Robustness and Stability: This refers to a method's ability to handle a diverse range of chemical systems and to converge reliably without manual intervention or failure.
- Usability and Practicality: Factors such as the quality of documentation, ease of installation and use, and the clarity of error messages significantly impact a researcher's ability to apply the tool effectively [5].
The Core Challenges in Design and Implementation
Even with a multi-dimensional view of performance, designing a benchmark that yields accurate, unbiased, and informative results is a non-trivial undertaking. Several core challenges must be navigated:
- The "Apples-to-Oranges" Comparison Problem: A critical challenge is ensuring that different software packages are performing the exact same scientific calculation. This goes beyond simply specifying a method (e.g., "DFT/B3LYP"). It requires meticulous control over parameters, algorithms, and numerical settings. As noted in a discussion on quantum chemistry benchmarks, it is surprisingly difficult to get different programs to do exactly the same calculation, with variations possible in aspects like quadrature grids, integration accuracy, and pseudopotential implementation [65]. A benchmark that fails to control these factors is comparing apples to oranges.
- Bias in Method and Dataset Selection: The choice of which methods to include and which datasets to use for testing can profoundly influence the outcome. A benchmark that includes a new method but only compares it against outdated or weak competitors will naturally show a favorable result. Similarly, selecting benchmark datasets that are unrepresentative of real-world challenges, or that happen to play to the strengths of a particular method, introduces subjectivity and bias [5]. A neutral benchmark should strive to be as comprehensive as possible, including all relevant methods and a variety of datasets that cover a wide chemical space [5] [14].
- Parameter Tuning and Software Configuration: A common pitfall, especially in benchmarks conducted by method developers, is extensively tuning the parameters of a new method while using only default settings for competing methods. This disadvantages the competitors and provides a biased representation of performance [5]. Furthermore, the use of different compiler flags or software versions can also lead to significant performance variations that are unrelated to the underlying algorithm's quality.
Table 1: Key Challenges in Computational Benchmarking and Their Implications
| Challenge | Description | Potential Consequence |
|---|---|---|
| Algorithmic & Parameter Parity | Ensuring identical physical approximations and numerical precision across software. | Comparisons reflect implementation details, not fundamental method quality. |
| Dataset Representativeness | Using datasets that are too small, simplistic, or chemically narrow. | Results do not generalize to real-world, complex research problems. |
| Resource Scaling | Measuring performance on a single hardware setup without considering parallel scaling. | Overlooks critical performance characteristics for large-scale problems on high-performance computing (HPC) systems. |
| Evaluation Metric Selection | Relying on a single metric (e.g., speed) without considering accuracy or robustness. | Selects for methods that are fast but inaccurate or unreliable. |
Essential Guidelines for Rigorous Benchmarking
To overcome these complexities, the computational science community has developed a set of best practices for designing, executing, and interpreting benchmarking studies. Adherence to these guidelines is essential for producing results that are trustworthy and useful for the broader research community.
Defining Purpose, Scope, and Neutrality
The first and most crucial step is to define the purpose and scope of the benchmark. Is it a "neutral" study conducted by an independent group to provide guidance to the community, or is it a study by method developers to demonstrate the merits of a new approach? Each has a place, but they have different requirements for comprehensiveness. A neutral benchmark should aim to include all available methods for a given type of analysis, while a developer-focused benchmark may compare against a representative subset of state-of-the-art and baseline methods [5]. In both cases, the key is to avoid bias. For a neutral benchmark, this means the research group should be equally familiar with all methods or collaborate with the original method authors to ensure each is evaluated under optimal conditions [5].
The Critical Role of Data and Evaluation Metrics
The selection of reference datasets and performance metrics forms the empirical foundation of any benchmark.
- Dataset Selection and Curation: A benchmark must utilize a variety of datasets to evaluate methods under a wide range of conditions. These can be real experimental data or simulated data with a known "ground truth." Simulated data is valuable for quantitatively measuring the ability to recover a known signal, but it must be shown to accurately reflect the properties of real data [5]. For real data, rigorous curation is essential. This includes standardizing chemical structures (e.g., using RDKit), neutralizing salts, removing duplicates, and identifying and handling experimental outliers to ensure data quality and consistency [14].
- Choosing Quantitative Performance Metrics: The selection of metrics must align with the scientific question. For predictive models, this could include R², mean absolute error, area under the receiver operating characteristic curve (AUC-ROC), or area under the precision-recall curve (AUC-PR) [19] [14]. It is often critical to evaluate performance both inside and outside a model's applicability domain to understand its limits [14]. Furthermore, computational performance metrics like wall-clock time, memory usage, and parallel scaling efficiency are necessary for a complete picture.
Table 2: Categories of Key Performance Indicators (KPIs) for Computational Chemistry Benchmarks
| KPI Category | Example Metrics | Relevance |
|---|---|---|
| Predictive Accuracy | R², Root-Mean-Square Error (RMSE), Balanced Accuracy, AUC-ROC, AUC-PR | Measures the scientific correctness and predictive power of the model's primary output. |
| Computational Performance | Time-to-solution (for energy, gradient, MD step), Memory (RAM) usage, Parallel Speedup/Efficiency | Measures the computational resource requirements and efficiency of the implementation. |
| Robustness & Stability | SCF Convergence Rate, Geometry Optimization Success Rate, Numerical Stability | Measures the reliability of the method across diverse chemical systems without manual intervention. |
| Practical Usability | Installation success rate, Quality of documentation, Runtime for standard test cases | Measures the "time to science" for an average researcher, impacting adoption. |
Standardized Protocols for Reproducibility
For a benchmark to be scientifically valid, it must be reproducible. This requires detailed reporting of experimental protocols.
- Software and Environment: Document exact software versions, compiler versions, and key library dependencies. The computational environment (operating system, CPU model, etc.) should also be specified.
- Methodology and Parameters: Every computational method must be defined with all relevant parameters explicitly listed. For a quantum chemistry benchmark, this includes the electronic structure method, basis set, convergence thresholds, and integration grids. For a molecular dynamics benchmark, this includes the force field, thermostat/barostat settings, and time step.
- Data and Code Availability: The highest standard of reproducibility is achieved by making the curated datasets and analysis code publicly available. This allows other researchers to verify results and build upon the work [14].
The following workflow diagram summarizes the key stages of a rigorous benchmarking process as discussed in this section.
Case Studies in Effective Benchmarking
Case Study 1: Predictive Modeling for Chemical Properties
A 2024 benchmarking study on tools for predicting toxicokinetic and physicochemical properties provides an exemplary model of rigorous design. The study selected twelve software tools to predict 17 different properties [14]. Its methodology directly addressed several core challenges:
- Data Curation: The authors collected 41 validation datasets from the literature and performed extensive curation. This included standardizing chemical structures, handling duplicates, and removing intra- and inter-dataset outliers to ensure data quality and consistency [14].
- Applicability Domain: The evaluation emphasized the performance of models inside their applicability domain, a crucial step for understanding real-world utility [14].
- Chemical Space Analysis: The researchers plotted their validation datasets against a reference chemical space (containing industrial chemicals, approved drugs, and natural products) to demonstrate the relevance and coverage of their benchmark, thereby increasing confidence in the evaluation's validity for real-life chemical categories [14].
This study moved beyond simple comparisons by identifying recurring best-performing tools and suggesting robust computational methods for high-throughput chemical assessment, providing valuable guidance to regulators and industry.
Case Study 2: High-Performance Computing and Scaling
While raw speed is a limited metric, computational efficiency remains vital, especially when considering large-scale problems. A key insight from computational researchers is that the most relevant test is often not speed on a single core, but scaling across many compute nodes [65]. This is because many researchers have access to large computing clusters, yet many electronic structure packages do not scale efficiently beyond a few nodes due to inherent algorithmic limitations or implementation choices. A robust benchmark for performance must, therefore, include scaling tests that measure how the time-to-solution decreases as more processors are added. A method that is moderately fast on a single core but exhibits near-ideal parallel scaling may be the most efficient choice for large molecules or long timescale molecular dynamics simulations, ultimately being more practical for cutting-edge research.
The Scientist's Toolkit: Essential Reagents for Benchmarking
To conduct a rigorous benchmarking analysis, researchers require a suite of tools and concepts. The following table details key "research reagents" for this field.
Table 3: Essential "Reagents" for Computational Benchmarking
| Tool / Concept | Function / Purpose | Example Implementations / Notes |
|---|---|---|
| Reference Datasets | Provides the ground truth for evaluating predictive accuracy and robustness. | Public databases (ChEMBL, PubChem), community benchmarks (MoleculeNet), or custom-simulated data with known properties. |
| Data Curation Pipeline | Standardizes and cleans molecular data to ensure consistency and remove errors. | In-house scripts using RDKit or CDK to standardize SMILES, remove duplicates, and neutralize salts [14]. |
| Performance Metrics | Quantifies different aspects of model and software performance. | R², RMSE, AUC-ROC (for classification), parallel speedup, memory footprint, convergence success rate. |
| Statistical Analysis | Determines if observed performance differences are meaningful and not due to random chance. | Wilcoxon signed-rank test for comparing method rankings, confidence interval estimation, regression analysis [19] [5]. |
| Reproducibility Framework | Ensures that all results can be independently verified. | Version-controlled code (Git), containerization (Docker/Singularity), workflow managers (Nextflow, Snakemake). |
| Chemical Space Visualization | Validates that benchmark datasets are representative of real-world problems. | Principal Component Analysis (PCA) applied to molecular fingerprints (e.g., ECFP, FCFP) [14]. |
The pursuit of faster computational tools is a noble and necessary driver of innovation in computational chemistry and drug discovery. However, this whitepaper has demonstrated that an exclusive or primary focus on speed tests is inherently limiting and can be scientifically counterproductive. The complexity of modern computational methods demands a more sophisticated approach to benchmarking. Effective benchmarking is a multifaceted, rigorous process that must be carefully designed to evaluate accuracy, robustness, scalability, and usability against well-defined scientific objectives using curated, representative data. By adopting the guidelines and protocols outlined here—defining a clear and neutral scope, meticulously selecting and curating data, employing a suite of quantitative metrics, and prioritizing reproducibility—researchers can produce benchmarks that truly illuminate the strengths and weaknesses of available tools. Such rigorous comparisons are not merely academic exercises; they are fundamental to building confidence in computational predictions, guiding the development of more powerful and reliable methods, and ultimately accelerating the pace of scientific discovery in the design of new drugs and materials.
In computational chemistry and drug discovery, robust model validation is not merely a final checkpoint but the foundational practice that distinguishes reliable, actionable research from potentially misleading advertisements. The core thesis of benchmarking in this field is to provide a rigorous, unbiased framework for assessing a model's true predictive power and practical utility in real-world scenarios, such as hit identification and lead optimization. Despite the increasing sophistication of machine learning and deep learning methods, many studies fail to prove genuine advances due to fundamental flaws in validation design. These include the use of inappropriate performance metrics, inadequate data splitting procedures, and a neglect for the quality of negative data, ultimately leading to models that perform well on benchmark datasets but fail in prospective drug discovery campaigns [19] [25]. This guide details the strategies—specifically, advanced cross-validation and the generation of high-quality negative data—essential for meaningful model validation, ensuring that computational tools genuinely accelerate scientific discovery.
Beyond Basic Cross-Validation: Strategies for Real-World Performance
Conventional random split cross-validation often produces optimistically biased performance estimates because test compounds are frequently structurally similar to those in the training set. This approach fails to assess a model's ability to generalize to truly novel chemical scaffolds, which is a primary goal in drug discovery. Advanced splitting strategies and evaluation metrics are required to simulate real-world application and provide a realistic picture of model performance.
Advanced Data Splitting Strategies
| Splitting Strategy | Core Principle | Simulated Real-World Scenario | Key Advantages |
|---|---|---|---|
| Scaffold Split [19] [66] | Splits data based on molecular Bemis-Murcko scaffolds. | Predicting activity for entirely new chemotypes. | Directly tests generalization to novel chemical series. |
| Time Split [66] | Uses older data for training and newer data for testing. | Mimics the use of historical data to predict future discoveries. | Accounts for temporal drift in data collection methods and chemical space. |
| k-fold n-Step Forward Cross-Validation (SFCV) [66] | Sorts data by a property (e.g., LogP) and sequentially expands training set. | Simulates the lead optimization process where properties are iteratively improved. | Tests model's ability to guide chemical space exploration towards more drug-like regions. |
Experimental Protocol for Sorted k-fold n-Step Forward Cross-Validation (SFCV):
- Dataset Preparation: Standardize molecular structures from SMILES using a toolkit like RDKit. Calculate a key physicochemical property such as LogP (partition coefficient) for each compound [66].
- Data Sorting and Binning: Sort the entire dataset by the calculated LogP value in descending order. Split the sorted list into
k(e.g., 10) sequential bins of equal size. - Iterative Training and Testing:
- Iteration 1: Use Bin 1 (highest LogP) as the training set. Use Bin 2 as the test set.
- Iteration 2: Combine Bins 1 and 2 as the new training set. Use Bin 3 as the test set.
- Continue this process until Bin
kis used as the test set.
- Model Evaluation: For each iteration, train the model and evaluate its performance on the test bin. The trend in performance across iterations indicates the model's utility in predicting progressively more drug-like compounds (typically with moderate LogP) [66].
Beyond AUC-ROC: A Multi-Metric Evaluation Framework
The Area Under the Receiver Operating Characteristic Curve (AUC-ROC) can be misleading for imbalanced datasets common in drug discovery, where inactive compounds vastly outnumber actives. A robust benchmarking thesis requires a suite of metrics [19] [50].
- Area Under the Precision-Recall Curve (AUC-PR): This metric should be used in conjunction with AUC-ROC, as it is more informative for imbalanced datasets. It directly answers the question: "Of all the compounds the model predicted as active, how many are truly active?" [19]
- Discovery Yield: This metric assesses a model's ability to enrich for active compounds in a prioritized list. It is calculated as the fraction of top-ranked compounds that are truly active, providing a direct measure of a model's value in a virtual screening campaign [66].
- Novelty Error: This measures a model's performance on compounds that are significantly different from its training data (its "applicability domain"). A high novelty error indicates poor generalization, a critical failure mode for practical use [66].
The Challenge of Negative Data: From Artifact to Asset
In virtual high-throughput screening (vHTS), "negative data" (confirmed inactive compounds) are as crucial as active compounds for training accurate models. However, the standard practice of using randomly selected compounds or artificially generated decoys as negatives introduces severe biases and artificially inflates performance metrics, rendering many validation studies unreliable [25].
Pitfalls of Low-Quality Negative Data
- Artificial Enrichment: Decoys are often chosen to be chemically dissimilar to actives, making them trivially easy for the model to distinguish. This results in over-optimistic performance that does not translate to real-world screening libraries where actives and inactives can be structurally similar [25] [50].
- Data Imbalance: The number of known inactives is typically very small compared to the vast number of untested compounds, which are often treated as negatives by default. This mislabeling noise confuses the model and reduces its predictive accuracy [25].
Strategies for Generating High-Quality Negative Data
Generating high-quality negative data requires no additional experimental work but demands careful computational design.
| Strategy | Methodology | Key Advantage |
|---|---|---|
| Ligand Randomization [25] | Dock known binders of Protein A into the structure of Protein B (where they are not known to bind). This uses experimental structures to create non-binders. | Generates negatives that are closely matched to positives in molecular properties, creating a challenging and realistic test. |
| Structural Isomer Generation [25] | Generate structural isomers of known active compounds using a tool like MAYGEN. These isomers have the same molecular formula but different connectivity and are unlikely to bind. | Creates negative data that is physically and chemically similar to actives, preventing models from relying on trivial property-based distinctions. |
Experimental Protocol for Generating Negative Data via Randomization and Isomerization:
- Curate Positive Protein-Ligand Pairs: Compile a set of known binding pairs from a database like PDBbind, where each entry is a protein structure (e.g., PDB ID 1FCX) and its cognate ligand [25].
- Generate Non-Binding Pairs via Cross-Docking:
- For a given protein structure (e.g., from 1FCX), prepare its binding site.
- Instead of using its native ligand, dock the ligand from a different, unrelated protein structure (e.g., from PDB ID 1G74) into the prepared site.
- A successfully docked pose of this "foreign" ligand constitutes a high-quality negative example, as it demonstrates the ligand can physically fit but is not a true binder [25].
- Generate Non-Binding Pairs via Isomerization:
- Select a known active ligand (e.g., from PDB ID 4QSW).
- Input its SMILES string into a structural isomer generator (e.g., MAYGEN) to produce a set of isomers.
- These isomers, which share the same atomic composition but different bond arrangements, serve as rigorous negative controls [25].
Integrating Strategies into a Coherent Benchmarking Framework
The ultimate goal is to integrate these strategies into an end-to-end validation pipeline that delivers a truthful assessment of model performance. Initiatives like the Critical Assessment of Computational Hit-finding Experiments (CACHE) exemplify this principle by running community-wide, blinded challenges where computational predictions are tested through rigorous experimental validation [67]. A robust benchmarking thesis must also account for the applicability domain of the models and the chemical space of the validation sets to ensure results are relevant to industrial chemicals, approved drugs, and other real-world chemical categories [14].
| Tool / Resource | Type | Primary Function in Validation | Example Use Case |
|---|---|---|---|
| RDKit [66] [14] | Open-Source Cheminformatics Library | Molecular standardization, fingerprint calculation (ECFP), and property calculation (LogP). | Featurizing molecules for model training; standardizing datasets from diverse sources. |
| ChEMBL [19] [50] | Public Bioactivity Database | Source of experimental bioactivity data for building training and benchmark sets. | Curating assays for virtual screening (VS) and lead optimization (LO) tasks. |
| PDBbind [25] [50] | Database of Protein-Ligand Complexes | Provides structured protein-ligand pairs for generating high-quality negative data. | Sourcing protein structures and their known binders for cross-docking experiments. |
| MAYGEN [25] | Structural Isomer Generator | Creates structural isomers of known actives to serve as challenging negative examples. | Generating non-binders that are chemically similar to active compounds. |
| Enamine REAL / ZINC [67] | Commercially Accessible Virtual Compound Libraries | Provides a source of purchasable compounds for prospective validation. | Procuring computationally predicted hits for experimental testing in blinded challenges. |
| ScaffoldSplitter (DeepChem) [66] | Software Function | Implements scaffold-based splitting of molecular datasets. | Ensuring training and test sets contain distinct chemotypes to test generalization. |
In computational chemistry, machine learning interatomic potentials (MLIPs) have emerged as a powerful tool to bridge the accuracy-cost gap between quantum mechanical methods and classical force fields. Conventional model validation has heavily relied on metrics like root-mean-square error (RMSE) or mean-absolute error (MAE) of energies and atomic forces on static test sets. However, evidence from recent benchmarking studies reveals a critical disconnect: MLIPs with excellent force accuracy often fail to reproduce physically correct dynamics in molecular simulations. This whitepaper examines the underlying causes of this discrepancy, synthesizes next-generation evaluation methodologies, and provides a framework for developing MLIPs robust enough for real-world scientific applications, particularly within the broader context of rigorous benchmarking in computational chemistry.
The primary goal of developing MLIPs is their deployment in Molecular Dynamics (MD) simulations to explore atomic-scale phenomena and predict macroscopic properties. The standard practice has been to use force and energy prediction errors on a held-out test set as a proxy for simulation quality [68]. This approach is increasingly revealed as insufficient.
State-of-the-art MLIPs, including Gaussian Approximation Potential (GAP), Neural Network Potential (NNP), and Deep Potential (DeePMD), frequently report remarkably low force errors, often below 0.1 eV Å⁻¹ [69]. Yet, when deployed in actual MD simulations, these models can exhibit pathological behaviors—from subtle inaccuracies in diffusion rates to catastrophic simulation collapse—that are not captured by static error metrics [68]. This paradox highlights a fundamental misalignment between the standard benchmarks and the practical objectives of atomistic modeling, underscoring a critical theme in model validation research: a good benchmark must test what the model is ultimately required to do.
Quantitative Evidence of the Disconnect
The gap between traditional metrics and simulation performance is not merely theoretical but is substantiated by consistent empirical findings across multiple studies and material systems.
Table 1: Documented Discrepancies Between Force Accuracy and Simulation Quality
| MLIP Model | Reported Force Error (eV Å⁻¹) | Observed Simulation Error/Discrepancy | Reference |
|---|---|---|---|
| Al MLIP (Botu et al.) | 0.03 MAE | 17% error in vacancy diffusion activation energy (0.1 eV vs DFT 0.59 eV) | [69] |
| Al MLIP (Vandermause et al.) | 0.05 RMSE (solid) | Discrepancies in surface adatom migration | [69] |
| Various MLIPs (GAP, NNP, SNAP, MTP) | 0.15 - 0.40 RMSE | 10-20% errors in vacancy formation energy and migration barrier | [69] |
| Si MLIPs (GAP, NNP, SNAP, MTP, DeePMD) | < 0.3 RMSE on vacancy test set | Poor performance on interstitial rare-event test set, energy bias of 10-13 meV/atom | [69] |
The data in Table 1 demonstrates that low average errors provide an incomplete picture. The failure to accurately predict properties like vacancy diffusion and migration barriers is particularly telling, as these are critical for understanding material behavior and are often the very targets of simulation campaigns.
Root Causes: Why Force Errors Are Misleading
The divergence between low force errors and poor simulation performance stems from several intrinsic properties of MLIPs and the nature of chemical systems.
The Rare Event Problem
Many critical physical processes, such as diffusion, phase transitions, and chemical reactions, are governed by rare events (REs). These involve atomic configurations that are high-energy states and thus have a low probability of being sampled in a standard training or testing set [69]. An MLIP can achieve low overall force errors while being highly inaccurate in these sparsely sampled but physically crucial regions of the potential energy surface (PES).
Non-Equilibrium and Defect Configurations
Similarly, defects (vacancies, interstitials, dislocations) and non-equilibrium structures are often underrepresented in datasets. The Si MLIP study showed that models trained on diverse datasets, including vacancies, performed well on vacancy testing but showed significant errors on interstitial testing, a configuration not included in training [69]. This highlights a key benchmarking challenge: ensuring out-of-distribution (OOD) generalizability.
The Dynamics of Error Propagation
In MD simulations, forces are integrated over time to update atomic positions and velocities. Small force errors, even if seemingly negligible at a single timestep, can accumulate over thousands of steps. This can lead to energy drift, where the total energy of the system is not conserved, or can push the system into unphysical regions of the PES where the MLIP makes wildly incorrect predictions, causing simulation "explosion" [68]. The stability of long-time-scale simulations is a metric that is entirely absent from static force-error evaluations.
Diagram 1: Dynamics of error propagation in MD simulations.
Next-Generation Benchmarking and Evaluation Metrics
Moving beyond force errors requires a new paradigm for benchmarking that directly assesses a model's performance in realistic application scenarios. The research community has begun to establish more robust frameworks.
Simulation-Centric Benchmarking
Novel benchmarks like those proposed by Fu et al. and LAMBench advocate for evaluating MLIPs based on their ability to reproduce macroscopic observables derived from MD trajectories [68] [10]. These observables connect directly to the scientific questions that motivate the simulations in the first place.
Table 2: Key Observables for Simulation-Centric Benchmarking
| System Type | Example Observables | Physical Property Probed |
|---|---|---|
| Water & Liquids | Radial Distribution Function (RDF), Diffusion Coefficient | Structure, dynamics, transport properties |
| Organic Molecules | Dihedral Angle Distributions, Mean-Squared Displacement (MSD) | Conformational preferences, flexibility |
| Peptides & Proteins | Root-Mean-Square Deviation (RMSD), Secondary Structure Stability | Folding, stability, function |
| Materials (Solid) | Virial Stress, Phonon Density of States, Defect Migration Barriers | Mechanical properties, thermal conductivity, kinetics |
Targeted Metrics for Rare Events and Defects
To specifically address the RE problem, researchers have developed targeted evaluation metrics. One approach is to create specialized testing sets, such as "RE-testing" sets, which consist of atomic configurations sampled from AIMD simulations during a rare event like a vacancy or interstitial migration [69]. The force errors are then calculated specifically for the atoms actively involved in the migration. This provides a more sensitive and relevant measure of model readiness for simulating dynamic processes than a global force RMSE.
The LAMBench Framework
The recently introduced LAMBench offers a comprehensive system for evaluating Large Atomistic Models (LAMs) along three critical axes [10]:
- Generalizability: Performance on data from diverse chemical domains and systems outside the training distribution.
- Adaptability: The model's capacity to be fine-tuned for specific downstream tasks.
- Applicability: The stability, efficiency, and conservativeness of the model in real-world simulations, such as MD.
This framework explicitly shifts the focus from isolated prediction accuracy to the holistic utility of a model as a tool for scientific discovery.
Diagram 2: Pillars of a comprehensive MLIP benchmarking framework.
A Protocol for Robust MLIP Development and Validation
Based on the identified challenges and new benchmarking approaches, the following protocol provides a pathway for developing and validating MLIPs for reliable use in real-world tasks.
Phase 1: Data Curation and Model Training
- Step 1: Diverse Training Set Generation. Actively include configurations relevant to the target application, such as transition states, defect structures, and non-equilibrium phases, going beyond simple bulk configurations [69].
- Step 2: Model Selection. Use initial force/energy RMSE as a preliminary filter, but not as the sole selection criterion.
Phase 2: Advanced Model Testing
- Step 3: RE-Based Evaluation. Calculate force and energy errors on dedicated rare-event and defect test sets (e.g.,
D_RE-VTestingfor vacancies,D_RE-ITestingfor interstitials) [69]. - Step 4: Property Prediction Test. Evaluate the model on easy-to-compute physical properties like elastic constants, energy-volume equations of state, and defect formation energies [69].
Phase 3: Simulation-Based Validation
- Step 5: Short MD Stability Test. Run multiple short MD simulations (e.g., 10-100 ps) from different initial conditions to check for catastrophic failures or energy drift [68].
- Step 6: Observable Comparison. Run longer, well-equilibrated MD simulations and compare key macroscopic observables (e.g., RDF, MSD, stress) against reference ab initio MD (AIMD) or experimental data [68].
- Step 7: Iterative Improvement. Use the insights from simulation failures and observable discrepancies to guide the augmentation of the training dataset or adjustment of model hyperparameters.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Tools and Resources for MLIP Benchmarking
| Tool/Resource | Type | Primary Function | Example/Reference |
|---|---|---|---|
| Reference Data | Dataset | Provides ground-truth energies/forces for training & testing | OMol25 [8], MPtrj [10] |
| RE Testing Sets | Custom Dataset | Evaluates model accuracy on critical migration paths | D_RE-VTesting, D_RE-ITesting [69] |
| MLIP Packages | Software | Implements various MLIP architectures for training & inference | GAP, DeePMD, MACE, NequIP [69] [68] |
| MD Engines | Software | Performs molecular dynamics simulations with MLIPs | LAMMPS, ASE, SchNetPack [68] |
| Benchmark Suites | Software Framework | Standardizes evaluation across models and tasks | LAMBench [10], Fu et al. suite [68] |
| Analysis Tools | Software | Computes observables from MD trajectories | MDANALYSIS, VMD, in-house scripts |
The reliance on low force errors as the primary validation metric for MLIPs is a dangerous oversimplification. True model reliability is demonstrated not by static prediction accuracy, but by the model's ability to enable physically correct and scientifically useful molecular simulations. The path forward requires a cultural and methodological shift in the computational chemistry community toward simulation-centric benchmarking. By adopting the frameworks and protocols outlined here—focusing on generalizability, stability, and the accurate reproduction of macroscopic observables—researchers can develop more robust and trustworthy MLIPs. This, in turn, will accelerate reliable scientific discovery in fields ranging from drug development to materials design.
Measuring Success: Validation Metrics, Statistical Techniques, and Comparative Model Analysis
In computational chemistry, the development of machine learning (ML) models and neural network potentials (NNPs) is accelerating the discovery of new molecules, materials, and drugs. However, the reliability of these models hinges on rigorous validation through systematic benchmarking. Benchmarking provides an objective framework for comparing model performance, guiding methodological improvements, and establishing trust in computational predictions for real-world scientific applications [10] [4]. The Critical Assessment of Structure Prediction (CASP) challenge, for instance, famously provided the rigorous benchmarking needed to advance protein structure prediction to the accuracy achieved by AlphaFold2 [10] [4].
At the heart of this benchmarking process are key validation metrics—Mean Absolute Error (MAE), Root Mean Square Error (RMSE), R-squared (R²), and Balanced Accuracy—which quantitatively assess a model's predictive quality. Properly interpreting these metrics allows researchers to select models that are not only statistically sound but also fit for purpose in specific chemical applications, from predicting molecular energies to classifying toxicological hazards [14].
Deep Dive into Core Validation Metrics
Metric Definitions and Mathematical Foundations
Table 1: Core Validation Metrics for Regression and Classification Models
| Metric | Mathematical Formula | Interpretation | Ideal Value | ||
|---|---|---|---|---|---|
| MAE | ( \frac{1}{n}\sum_{i=1}^{n} | yi-\hat{y}i | ) | Average magnitude of absolute errors, robust to outliers. | Closer to 0 |
| RMSE | ( \sqrt{\frac{1}{n}\sum{i=1}^{n}(yi-\hat{y}_i)^2} ) | Average magnitude of errors, penalizes large errors more heavily. | Closer to 0 | ||
| R² | ( 1 - \frac{\sum{i=1}^{n}(yi-\hat{y}i)^2}{\sum{i=1}^{n}(y_i-\bar{y})^2} ) | Proportion of variance in the dependent variable that is predictable from the independent variables. | Closer to 1 | ||
| Balanced Accuracy | ( \frac{1}{2}\left(\frac{TP}{TP+FN} + \frac{TN}{TN+FP}\right) ) | Average of sensitivity and specificity, suitable for imbalanced datasets. | Closer to 1 |
Comparative Analysis and Selection Guidelines
Each metric provides a distinct perspective on model performance, and their combined interpretation is crucial.
MAE vs. RMSE: Both measure average prediction error, but RMSE's squaring of the error term gives it greater sensitivity to large errors and outliers. In contexts where large errors are particularly undesirable, such as predicting the energy of an unstable molecular configuration, a higher RMSE relative to MAE signals the presence of consequential, large errors that MAE might downplay [70]. MAE, being a linear score, is often favored for its straightforward interpretability as the average error [70].
R² (Coefficient of Determination): R² explains the proportion of variance in the target variable that is captured by the model. However, it must be used with caution. It can be deceptive when applied to nonlinear models, and a high R² does not automatically mean the model is correct, especially if there is systematic error [70]. Furthermore, it is most reliable for linear relationships and can be misleading in complex, high-dimensional chemical spaces.
Balanced Accuracy: This metric is the go-to choice for classification tasks with imbalanced datasets, such as predicting whether a compound is toxic when the majority of compounds in the dataset are non-toxic. By averaging the accuracy of both classes, it prevents a model from achieving artificially high performance by simply always predicting the majority class [14].
Table 2: Key Characteristics and Guidelines for Metric Selection
| Metric | Primary Use Case | Strengths | Limitations & Cautions |
|---|---|---|---|
| MAE | General-purpose error measurement; robust reporting. | Intuitive, easy to interpret; robust to outliers. | Does not penalize large errors heavily. |
| RMSE | When large errors are particularly undesirable. | Sensitive to large errors/variance; same units as target. | Highly sensitive to outliers; can be dominated by a few bad predictions. |
| R² | Explaining model performance relative to a simple mean. | Standardized, scale-independent interpretation. | Can be misleading for non-linear models; sensitive to outliers [70]. |
| Balanced Accuracy | Classification with imbalanced datasets. | Provides a realistic performance measure on imbalanced data. | Not suitable for regression; requires careful threshold setting. |
Experimental Protocols for Model Validation
A Standard Workflow for Benchmarking in Computational Chemistry
Robust model validation requires more than just calculating final metrics; it demands a rigorous experimental protocol. The following workflow, formalized by benchmarks like LAMBench, outlines the key steps for a comprehensive evaluation [10].
Protocol Details and Best Practices
Data Curation and Preprocessing: The foundation of any reliable model is high-quality data. This involves standardizing molecular representations (e.g., SMILES), normalizing feature scales, and applying dimensionality reduction techniques like Principal Component Analysis (PCA) to handle high-dimensional spectral data [71]. Outlier detection, for instance using Cook's Distance, is critical to remove influential data points that could distort the regression model [71].
Data Splitting and Cross-Validation: To obtain a realistic estimate of model performance on unseen data, the curated dataset must be split. A standard practice is to use a holdout test set, which is only used for the final evaluation. For model development and tuning, k-fold cross-validation (e.g., k=3) is employed, where the model is trained and validated on different subsets of the training data to ensure performance is consistent and not due to a particular random split [71].
Model Evaluation and Metric Calculation: With the data prepared and models trained, the next step is the quantitative evaluation on the test set. This involves calculating the suite of metrics—MAE, RMSE, R² for regression, and Balanced Accuracy for classification—as defined in Section 2. A study predicting drug release from coated formulations, for example, successfully used R², RMSE, and MAE to demonstrate a Multilayer Perceptron (MLP) model's superior performance (R²=0.9989) over linear models [71].
Performance Analysis and Interpretation: The final, crucial step is to synthesize the quantitative results with complementary graphical techniques. Parity plots, which compare predicted vs. actual values, and residual plots are essential for diagnosing model behavior—such as identifying systematic biases—that purely numerical indicators may overlook [70] [71]. This holistic analysis ensures that a model is not only statistically accurate but also chemically and physically sound before it is selected for deployment.
Table 3: Key Tools and Resources for Computational Chemistry Benchmarking
| Tool / Resource | Type | Primary Function | Relevance to Benchmarking |
|---|---|---|---|
| OMol25 Dataset [15] [72] | Dataset | Massive, diverse dataset of molecular simulations for training MLIPs. | Provides a high-quality, standardized foundation for training and testing models, enabling fair comparisons. |
| LAMBench [10] | Benchmarking Platform | System to evaluate Large Atomistic Models (LAMs). | Offers a standardized set of tasks to assess model generalizability, adaptability, and applicability. |
| MOFSimBench [13] | Specialized Benchmark | Evaluates models on Metal-Organic Frameworks (MOFs). | Provides domain-specific validation for tasks like structure optimization and host-guest interactions. |
| Curve Fitting Toolbox (MATLAB) [73] | Software Toolbox | Fits and evaluates regression models, computes metrics. | Aids in model development, hyperparameter tuning, and calculation of R², RMSE, and MAE. |
| Applicability Domain (AD) [14] | Methodology | Defines the chemical space a model is reliable for. | Critical for interpreting metrics; a model's high performance is only valid within its AD. |
The rigorous validation of computational chemistry models using MAE, RMSE, R², and Balanced Accuracy is not a mere procedural step but the cornerstone of scientific credibility in the field. As the development of large atomistic models and AI-driven discovery tools progresses, the frameworks for benchmarking them must evolve in parallel [10] [4]. A profound understanding of these metrics—their calculations, nuances, and appropriate contexts—empowers researchers to critically evaluate new methods, foster replicable and transparent science, and ultimately accelerate the development of reliable in-silico tools for designing the next generation of medicines, materials, and sustainable technologies.
In computational chemistry, the validation of models against reliable benchmarks is the cornerstone of scientific progress and reliability. Benchmarking provides the critical framework for assessing the performance, accuracy, and generalizability of computational models, from density functional theory (DFT) functionals to modern machine learning potentials. The ultimate goal is to develop models that serve as robust approximations of the universal potential energy surface (PES), which is defined by the first-principles solutions to the Schrödinger equation under the Born-Oppenheimer approximation [10]. The journey toward this goal, however, is fragmented by domain-specific challenges, including the use of different exchange-correlation functionals, basis sets, and pseudopotentials across research fields, which complicates the creation of a unified benchmark [10].
The process of benchmarking is not merely about achieving high accuracy on static test sets. It must also evaluate a model's performance in real-world application scenarios, ensuring predictions are not only numerically accurate but also physically meaningful and stable in dynamic simulations [10]. This technical guide details the core statistical techniques—specifically confidence intervals and regression analysis—that underpin robust model comparison within this context. These methods are essential for quantifying uncertainty, enabling meaningful comparisons between methodologies, and ultimately fostering the development of more reliable and universal atomistic models.
Foundational Concepts in Model Validation
Before delving into statistical techniques, it is crucial to establish the core objectives of model validation in computational chemistry. Benchmarks like the recently introduced LAMBench are designed to evaluate Large Atomistic Models (LAMs) across three fundamental capabilities [10]:
- Generalizability: The accuracy of a model on data that shares the same distribution as its training set (in-distribution) and, more challengingly, on data from different distributions (out-of-distribution).
- Adaptability: The model's capacity to be fine-tuned for tasks beyond direct potential energy prediction, such as structure-property relationship tasks.
- Applicability: The stability and efficiency of the model when deployed in practical simulations, such as molecular dynamics, where properties like energy conservation are critical [10].
A significant challenge in the field, as identified in drug discovery contexts, is the use of overly simplistic benchmarking methodologies. For instance, the traditional approach of multiplying phase transition success rates to estimate a drug's overall probability of success (POS) has been shown to systematically overestimate success and underestimate risk [29]. This highlights a broader issue: without robust statistical evaluation, benchmarks can provide a false sense of model accuracy and utility. The community is therefore moving towards more dynamic and nuanced benchmarking approaches that leverage large, harmonized, and continuously updated datasets to provide a more accurate view of success and risk [29].
Confidence Intervals for Robust Performance Estimation
In benchmarking, a single performance metric (e.g., a mean absolute error) is an incomplete picture. It provides a point estimate but fails to convey the precision or reliability of that estimate. Confidence intervals are a fundamental statistical tool that quantify the uncertainty around a point estimate, providing a range of plausible values for the true population parameter.
Methodology and Application
The construction of a confidence interval for a mean prediction error, for example, typically relies on the standard error of the mean. For a dataset of n independent observations, the (1 - α)% confidence interval is calculated as:
CI = x̄ ± t*(α/2, n-1) * (s / √n)
Where x̄ is the sample mean error, s is the sample standard deviation, and t*(α/2, n-1) is the critical value from the t-distribution with n-1 degrees of freedom.
In computational chemistry, this is applied to evaluate model performance on a benchmark dataset. Consider a benchmark that calculates the root-mean-square error (RMSE) of forces predicted by a model across a diverse set of molecular configurations. Reporting the RMSE alongside its confidence interval allows researchers to assess not just the model's average accuracy, but also the stability of that performance. A narrow confidence interval indicates high confidence in the reported accuracy, while a wide interval suggests the estimate is less precise, potentially due to a small test set or high variance in model performance across different system types. This is crucial for determining if one model is truly superior to another or if observed differences could be due to random chance.
Experimental Protocol for Confidence Interval Calculation
- Run Inference: Execute the model(s) under evaluation on the designated benchmark test set to generate predictions (e.g., energies, forces) for each data point.
- Calculate Errors: Compute the error for each data point as the difference between the model's prediction and the reference value (e.g., high-level DFT calculation).
- Compute Summary Statistics: Calculate the mean error (
x̄) and standard deviation of the errors (s) for the specific metric of interest across thensamples in the test set. - Determine Critical Value: Select the confidence level (e.g., 95%) and find the corresponding t-distribution critical value
t*(0.025, n-1). - Calculate Interval: Compute the margin of error and add/subtract it from the sample mean to establish the upper and lower bounds of the confidence interval.
- Report Results: Present the point estimate (e.g., mean RMSE) alongside the confidence interval in all reports and publications.
Regression Analysis for Method Comparison
Regression analysis is a powerful statistical technique for quantifying the relationship between variables. In model benchmarking, it is extensively used for method comparison, such as assessing the agreement between a new computational method and a high-level theory or experimental reference standard.
Linear Regression for Assessing Agreement
Ordinary Least Squares (OLS) regression is a standard approach. The linear model Y = β₀ + β₁X + ε is fitted, where Y is the predicted value from the model under test, X is the reference value, β₀ is the intercept, β₁ is the slope, and ε is the error term. The results are interpreted as follows:
- Ideal Agreement: A perfect agreement would result in a regression line with an intercept (
β₀) of zero and a slope (β₁) of one. - Systematic Bias: A non-zero intercept indicates a constant additive bias in the predictions. A slope different from one indicates a proportional scaling bias.
- Coefficient of Determination (R²): This metric, often used in benchmarks like stock prediction algorithms [74], indicates the proportion of variance in the reference data that is explained by the model. A value close to 1.0 indicates strong predictive power.
The following table summarizes key regression metrics and their interpretation in a benchmarking context.
Table 1: Key Regression Metrics for Benchmarking Computational Chemistry Models
| Metric | Formula | Interpretation in Benchmarking | Ideal Value |
|---|---|---|---|
| Slope (β₁) | Cov(X,Y) / Var(X) | Measures proportional bias of the model. | 1.0 |
| Intercept (β₀) | Ȳ - β₁X̄ | Measures constant additive bias of the model. | 0.0 |
| R-squared (R²) | 1 - [SS~res~ / SS~tot~] | Proportion of variance in reference data explained by the model. | 1.0 |
| Root Mean Square Error (RMSE) | √[ Σ(Y~i~ - X~i~)² / n ] | Average magnitude of prediction error, in the units of the data. | 0.0 |
Advanced Regression Techniques
For more complex relationships, other forms of regression are valuable:
- Weighted Least Squares (WLS): Used when the variance of the errors is not constant (heteroscedasticity). WLS assigns a weight to each data point, often inversely proportional to the variance of its error, to provide a more efficient estimate. This is particularly relevant in multi-fidelity benchmarking where data from different levels of theory (e.g., PBE vs. hybrid functionals) are combined [10].
- Deming Regression: A standard OLS assumes the reference values
Xare measured without error. In computational chemistry, even reference DFT calculations have inherent errors. Deming regression accounts for errors in both theXandYvariables, providing a more realistic assessment of the relationship.
Experimental Protocol for Regression-Based Benchmarking
- Data Collection: Gather paired data
(X_i, Y_i)whereX_iis the reference value (e.g., ωB97M-V/def2-TZVPD energy [15]) andY_iis the value predicted by the model under test. - Data Visualization: Create a scatter plot of
Yvs.Xwith a line of perfect agreement (Y=X). This provides an initial visual assessment of bias and outliers. - Model Fitting: Perform an OLS regression of
YonXto obtain the estimated slope, intercept, and R² value. - Residual Analysis: Plot the residuals (Y - Ŷ) against the predicted values (Ŷ). A random scatter indicates a good fit; any pattern suggests model misspecification (e.g., non-linearity, heteroscedasticity).
- Hypothesis Testing: Conduct statistical tests (e.g., t-tests) to determine if the intercept and slope are significantly different from 0 and 1, respectively. A significant p-value (e.g., < 0.05) indicates a statistically significant bias.
- Reporting: Report the regression parameters, their confidence intervals, and the R² value. The scatter plot with the regression line and the line of perfect agreement should be included for clarity.
An Integrated Workflow for Robust Model Validation
The statistical techniques described above are most powerful when integrated into a systematic benchmarking workflow. The following diagram visualizes this multi-stage process, from initial model evaluation to final statistical comparison and interpretation.
Diagram 1: A sequential workflow for robust model validation, integrating confidence intervals and regression analysis.
Successful benchmarking relies on a combination of software, datasets, and computational resources. The table below details key components of the modern computational chemist's toolkit for rigorous model validation.
Table 2: Research Reagent Solutions for Computational Chemistry Benchmarking
| Item Name | Type | Function / Application | Example / Reference |
|---|---|---|---|
| LAMBench | Benchmarking Platform | A comprehensive system for evaluating Large Atomistic Models (LAMs) on generalizability, adaptability, and applicability [10]. | LAMBench GitHub |
| High-Accuracy Datasets | Data | Large, diverse datasets of quantum chemical calculations used for training and, crucially, testing model performance. | Open Molecules 2025 (OMol25) [15] |
| Neural Network Potentials (NNPs) | Software/Model | Machine learning models trained to approximate potential energy surfaces; the primary objects of benchmarking. | eSEN, UMA models [15] |
| Dynamic Benchmarking Tools | Software/Methodology | Advanced tools that use continuously updated data and refined methodologies to avoid over-optimism in success rates. | Intelligencia AI's Dynamic Benchmarks [29] |
| Statistical Computing Environment | Software | An environment for performing regression analysis, calculating confidence intervals, and generating statistical plots. | R, Python (with scipy, statsmodels, scikit-learn) |
| Reference Quantum Chemistry Code | Software | Software used to generate high-accuracy reference data against which new models are benchmarked. | Codes that run ωB97M-V/def2-TZVPD [15] |
The rigorous validation of computational chemistry models through benchmarking is a statistical endeavor as much as a scientific one. Confidence intervals and regression analysis provide the formal framework to move beyond qualitative comparisons, enabling researchers to quantify uncertainty, identify systematic biases, and make statistically sound claims about model performance. As the field progresses towards the goal of universal, ready-to-use atomistic models, the adoption of these robust statistical techniques will be paramount. They ensure that progress is measured reliably and that the models driving scientific discovery are not just accurate on paper, but are also trustworthy and applicable in practice.
Benchmarking serves as the cornerstone of validation in computational chemistry, providing a rigorous framework for evaluating the performance, accuracy, and applicability of various modeling approaches. As the field evolves with an influx of complex algorithms and increasing computational power, systematic benchmarking has become indispensable for guiding researchers in selecting appropriate methods for specific scientific inquiries. This comparative analysis examines three foundational methodologies—Quantitative Structure-Activity Relationship (QSAR) models, Neural Network Potentials (NNPs), and Density Functional Theory (DFT)—within the context of modern benchmarking practices. The critical importance of benchmarking is highlighted by recent paradigm shifts in validation metrics, where traditional measures like balanced accuracy are being reconsidered in favor of positive predictive value (PPV) for specific applications such as virtual screening, reflecting the field's maturation toward context-dependent method assessment [51].
Each method occupies a distinct niche in the computational ecosystem: QSAR models excel at leveraging chemical patterns from existing bioactivity data; NNPs bridge the accuracy-efficiency gap between quantum mechanics and classical force fields; and DFT provides a first-principles reference standard, albeit at high computational cost. By examining these methods through a benchmarking lens, this review aims to equip researchers with the critical framework necessary for selecting and validating computational approaches tailored to their specific research objectives, particularly in drug discovery and materials science where prediction reliability directly impacts experimental success and resource allocation.
Methodological Foundations
QSAR Models: Ligand-Based Predictive Modeling
Quantitative Structure-Activity Relationship (QSAR) modeling represents a cornerstone of computer-assisted drug discovery, with a history spanning over six decades. These models establish mathematical relationships between molecular descriptors (quantitative representations of chemical structures) and biological activities to predict properties of novel compounds. Traditional QSAR approaches, including Multiple Linear Regression (MLR) and Partial Least Squares (PLS), generate linear equations correlating structural features with bioactivity [75]. However, contemporary best practices have evolved to address modern challenges, particularly with the exponential growth of chemical libraries and the shift from lead optimization to virtual screening applications [51].
The fundamental principle underlying QSAR is that chemically similar compounds exhibit similar biological activities. Molecular structures are typically encoded using descriptors such as extended connectivity fingerprints (ECFPs) or functional-class fingerprints (FCFPs), which capture topological and pharmacophoric features, respectively [75]. For example, ECFPs are generated by systematically recording the neighborhood of each non-hydrogen atom into multiple circular layers up to a given diameter, then mapping these atom-centered sub-structural features into integer codes that form the fingerprint [75]. Recent methodological advances have emphasized robust validation protocols, applicability domain assessment, and appropriate metric selection based on the specific context of use, moving beyond traditional practices to address the realities of imbalanced chemical datasets [51].
Neural Network Potentials: Machine-Learned Force Fields
Neural Network Potentials (NNPs) represent an innovative fusion of machine learning and molecular simulation, designed to achieve Density Functional Theory (DFT)-level accuracy at a fraction of the computational cost. These potentials train neural networks on reference quantum mechanical data to predict potential energy surfaces and atomic forces, enabling large-scale molecular dynamics simulations that would be prohibitively expensive with direct quantum mechanical methods [76]. The recent emergence of massive datasets like Open Molecules 2025 (OMol25)—containing over 100 million molecular snapshots with DFT-calculated properties—has dramatically accelerated NNP development by providing unprecedented training data across diverse chemical spaces [6].
Architecturally, NNPs like the Deep Potential (DP) scheme incorporate physical symmetries such as translation, rotation, and periodicity to ensure conservation laws are obeyed [76]. More recent advances address a key limitation of early NNPs—their difficulty modeling long-range intermolecular interactions—by explicitly incorporating electrostatic and dispersion corrections. For instance, the CombineNet model integrates a machine learning-based charge equilibration scheme for electrostatics and the Machine-Learning eXchange-hole Dipole Moment (MLXDM) model for dispersion, significantly improving accuracy for non-covalent interactions [77]. Frameworks such as EMFF-2025 demonstrate how transfer learning strategies can leverage pre-trained models on specialized chemical systems (e.g., high-energy materials) to create versatile potentials with minimal additional training data [76].
Density Functional Theory: The Quantum Mechanical Benchmark
Density Functional Theory (DFT) stands as the most widely used quantum mechanical method for materials science and chemistry, providing a first-principles approach to electronic structure calculation without empirical parameters. By solving for the electron density rather than individual wavefunctions, DFT achieves favorable scaling (typically O(N³) for system size N) compared to more accurate wavefunction-based methods like coupled cluster theory, which can scale as O(N⁷) or higher [6]. This compromise between accuracy and computational feasibility makes DFT the reference standard for training machine learning potentials and validating empirical methods.
Modern DFT calculations employ sophisticated exchange-correlation functionals (e.g., ωB97M-V) and basis sets (e.g., def2-TZVPD) to achieve chemical accuracy across diverse molecular systems [6] [8]. The fundamental Hohenberg-Kohn theorems establish that all ground-state properties are functionals of the electron density, while the Kohn-Sham approach constructs a fictitious system of non-interacting electrons that reproduces the same density. Despite its widespread success, DFT faces limitations in describing van der Waals interactions, strongly correlated systems, and reaction barriers, necessitating careful functional selection for specific applications. Nonetheless, its role as the computational foundation for both NNPs (as training data source) and QSAR models (as descriptor generator) underscores its central position in the computational chemistry ecosystem.
Performance Benchmarking and Comparative Analysis
Quantitative Performance Metrics Across Methods
Table 1: Performance Comparison Across Computational Methods
| Method | Accuracy Domain | Computational Cost | Training Data Requirements | Key Performance Metrics |
|---|---|---|---|---|
| QSAR (DNN) | Hit prediction efficiency, ADMET properties [78] | Low (seconds per prediction) | 6069 compounds for r² ~0.90 [75] | PPV: ~30% higher hit rate than balanced models [51], R²pred > 0.60 [75] |
| QSAR (RF) | Virtual screening, bioactivity classification [75] | Low to moderate | 6069 compounds for r² ~0.90 [75] | Balanced accuracy, R²pred ~0.84 with limited data [75] |
| NNPs (EMFF-2025) | Energetics, mechanical properties, reaction mechanisms [76] | Moderate (DFT: 10,000× faster) [6] | Transfer learning with minimal additional data [76] | MAE: 0.59 kcal/mol (energy), MAE: <2 eV/Å (forces) [76] |
| NNPs (OMol25-trained) | Reduction potentials, electron affinities [8] | Moderate to high | 100M+ DFT calculations [6] | MAE: 0.262V (organometallic reduction potentials) [8] |
| DFT (ωB97M-V/def2-TZVPD) | Electronic structure, reaction energies [6] | High (reference standard) | Not applicable | Chemical accuracy (1-3 kcal/mol), CCSD(T)/CBS benchmark [6] |
Table 2: Specialized Application Performance
| Application Domain | Best Performing Method | Key Results | Limitations |
|---|---|---|---|
| Virtual Screening (Hit Identification) | QSAR with high PPV on imbalanced datasets [51] | 30% more true positives in top predictions vs balanced models [51] | Requires large, diverse training sets; applicability domain critical |
| Energetic Materials Property Prediction | EMFF-2025 NNP [76] | Predicts structure, mechanical properties, decomposition of 20 HEMs with DFT accuracy [76] | Limited to C, H, N, O elements; training set must cover full intermolecular distance range [76] |
| Reduction Potential Prediction | UMA-S NNP (organometallic); B97-3c DFT (main-group) [8] | MAE 0.262V (organometallics); MAE 0.260V (main-group) [8] | NNPs less accurate for main-group reduction potentials (MAE 0.261-0.505V) [8] |
| Electron Affinity Prediction | OMol25-trained NNPs competitive with DFT [8] | Comparable or better than low-cost DFT methods despite no explicit physics [8] | Bond breaking issues upon electron addition for some methods [8] |
Critical Insights from Method Comparison
The benchmarking data reveals a complex performance landscape where method superiority is highly context-dependent. For virtual screening applications where experimental validation is limited to small compound batches (e.g., 128 molecules per plate), QSAR models optimized for positive predictive value (PPV) significantly outperform those maximizing balanced accuracy, demonstrating approximately 30% higher true positive rates in top-ranked predictions [51]. This finding underscores the importance of aligning validation metrics with practical constraints rather than relying on traditional statistical measures.
Neural Network Potentials demonstrate remarkable capability in bridging the accuracy-efficiency gap, with EMFF-2025 achieving near-DFT accuracy for energetic materials while enabling simulations thousands of times faster than direct quantum mechanical calculations [76]. Surprisingly, OMol25-trained NNPs perform comparably to or even exceed low-cost DFT methods for predicting charge-related properties like reduction potentials and electron affinities, despite not explicitly incorporating Coulombic physics in their architecture [8]. This counterintuitive result suggests that data volume and diversity can potentially compensate for explicit physical modeling in certain applications.
The benchmarking results also reveal important method-specific limitations. Traditional QSAR methods like MLR demonstrate significant overfitting with small training sets, yielding R²pred values near zero despite high training set correlation [75]. NNPs face challenges in describing long-range interactions without explicit corrections [77], while DFT methods struggle with computational cost that scales dramatically with system size, making biologically relevant systems often prohibitively expensive [6].
Integrated Workflows and Experimental Protocols
Virtual Screening Workflow Using QSAR Models
Diagram 1: QSAR Virtual Screening Workflow. This protocol emphasizes PPV-driven hit selection for experimental validation, incorporating scaffold-aware splitting and applicability domain assessment [75] [51].
Detailed Protocol for QSAR-Based Virtual Screening:
Data Curation and Preparation: Collect bioactivity data from public databases (ChEMBL, PubChem) or high-throughput screening campaigns. For a TNBC inhibitor discovery campaign, 7,130 molecules with reported MDA-MB-231 inhibitory activities were assembled from ChEMBL [75].
Molecular Standardization and Descriptor Generation: Standardize molecular structures (tautomer standardization, charge normalization) and generate molecular descriptors. The software devised 613 descriptors from AlogP_count, ECFP, and FCFP for model generation [75]. ECFP are circular topological fingerprints generated by systematically recording the neighborhood of each non-hydrogen atom into multiple circular layers.
Dataset Splitting: Implement scaffold-aware or cluster-aware splitting to ensure structural diversity between training and test sets, avoiding artificial inflation of performance metrics. The ProQSAR framework formalizes this process with reproducible splitting protocols [79].
Model Training and Validation: Train multiple algorithm types (DNN, RF, PLS, MLR) using the training set. For the TNBC dataset, models were trained on three different set sizes (6069, 3035, and 303 compounds) and validated on a fixed test set of 1,061 compounds [75]. Critical metrics include PPV for virtual screening applications and R²pred for regression tasks.
Virtual Screening and Hit Selection: Apply the validated model to screen ultra-large chemical libraries (e.g., Enamine REAL Space). Select top-ranking compounds based on PPV-optimized rankings rather than raw prediction scores, focusing on batches sized for experimental constraints (e.g., 128 compounds for 1536-well plates) [51].
NNP Development and Validation Workflow
Diagram 2: NNP Development Workflow. This protocol highlights transfer learning from large pretrained models and explicit incorporation of long-range interactions [77] [6] [76].
Detailed Protocol for NNP Development:
Reference Data Generation: Perform high-level DFT calculations (e.g., ωB97M-V/def2-TZVPD) on diverse molecular configurations. The OMol25 dataset utilized six billion CPU hours to generate over 100 million molecular snapshots with up to 350 atoms, including biomolecules, electrolytes, and metal complexes [6].
Architecture Selection and Pretraining: Select appropriate NNP architecture (e.g., Deep Potential, eSEN, UMA) and pretrain on large datasets. For EMFF-2025, a pre-trained DP-CHNO-2024 model served as the foundation, leveraging transfer learning to minimize required training data [76].
Transfer Learning and Specialization: Fine-tune the pre-trained model on domain-specific data. EMFF-2025 incorporated minimal additional training data specific to high-energy materials while maintaining generalizability across C, H, N, O systems [76].
Model Validation: Validate against DFT references for energies and forces. EMFF-2025 achieved mean absolute errors (MAE) predominantly within ±0.1 eV/atom for energy and ±2 eV/Å for forces across 20 high-energy materials [76].
Explicit Long-range Corrections: Incorporate machine learning-based charge equilibration schemes for electrostatics and MLXDM for dispersion interactions to address the limitation of local atomic environment descriptions [77].
Experimental Benchmarking: Validate model predictions against experimental data for mechanical properties, thermal decomposition behavior, and electronic properties. EMFF-2025 successfully predicted structure, mechanical properties, and decomposition characteristics of 20 high-energy materials, benchmarking against experimental data [76].
Table 3: Essential Computational Tools and Resources
| Tool/Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| OMol25 Dataset [6] | Reference Dataset | 100M+ DFT-calculated molecular snapshots for training NNPs | Provides foundational training data for general-purpose NNPs across diverse chemistry |
| ProQSAR [79] | Modeling Framework | Modular, reproducible QSAR development with standardized validation | Formalizes end-to-end QSAR development with versioned artifacts and audit reports |
| VEGA [80] | QSAR Platform | Integrated (Q)SAR models for environmental property prediction | Persistence, bioaccumulation, and mobility assessment of cosmetic ingredients |
| ECFP/FCFP [75] | Molecular Descriptors | Circular topological fingerprints capturing structural features | Standard molecular representations for QSAR and machine learning models |
| Deep Potential (DP) [76] | NNP Architecture | Neural network potential framework incorporating physical symmetries | Molecular dynamics simulations with DFT-level accuracy for complex systems |
| EMFF-2025 [76] | Specialized NNP | Pre-trained potential for energetic materials containing C, H, N, O | Prediction of mechanical properties and decomposition mechanisms of HEMs |
| B97-3c Functional [8] | DFT Method | Low-cost composite density functional with minimal basis set | Balanced accuracy/efficiency for reduction potential and electron affinity prediction |
| Applicability Domain Assessment [80] | Validation Tool | Identifies compounds outside model's reliable prediction space | Critical for QSAR model interpretation and reliable virtual screening |
This comparative analysis demonstrates that benchmarking in computational chemistry has evolved from simple accuracy comparisons to context-dependent validation frameworks that consider practical application constraints. The paradigm shift in QSAR modeling from balanced accuracy to PPV optimization for virtual screening reflects this maturation, acknowledging that real-world utility depends on specific use contexts rather than universal performance metrics [51]. Similarly, the surprising performance of NNPs on charge-based properties despite lacking explicit physics highlights how data volume and diversity can sometimes compensate for methodological limitations [8].
Future methodological development will likely focus on hybrid approaches that leverage the respective strengths of each methodology. NNPs trained on massive DFT datasets already demonstrate remarkable transferability, while QSAR models optimized for specific decision contexts offer practical utility in drug discovery pipelines. The emergence of standardized frameworks like ProQSAR [79] and benchmark datasets like OMol25 [6] represents significant progress toward reproducible, comparable model assessment. As computational chemistry continues to integrate machine learning approaches, robust benchmarking practices will remain essential for translating methodological advances into practical scientific discoveries across drug development, materials design, and environmental chemistry.
Benchmarking is a critical practice in computational chemistry research for validating model performance, establishing trust in predictions, and guiding the selection of appropriate methods for specific scientific questions. In the context of neural network potentials (NNPs), rigorous benchmarking ensures these machine-learned models can reliably replace or supplement traditional computational methods like density functional theory (DFT) and coupled cluster theory [15]. This case study focuses specifically on evaluating NNP performance for predicting charge-related molecular properties—a particularly challenging domain due to the complex electron transfer processes and long-range interactions involved.
Charge-related properties such as electron affinity and reduction potential are essential in numerous chemical applications, from designing battery materials to predicting drug behavior. However, many modern NNPs lack explicit physics-based treatment of charge interactions, raising questions about their ability to accurately model these sensitive properties [81] [82]. This analysis examines how the computational chemistry community benchmarks NNPs against experimental data and high-level theoretical methods to validate their performance, with particular attention to the groundbreaking Open Molecules 2025 (OMol25) dataset and associated models [15].
Experimental Benchmarking Methodologies
Cross-Methodological Validation Frameworks
Benchmarking NNPs for charge-related properties requires multiple validation strategies to assess different aspects of model performance. Researchers employ several complementary approaches:
Experimental Comparison: Direct comparison against experimentally measured properties provides the most reliable validation. For electron affinity, this involves comparing predicted versus measured energy differences between neutral molecules and their anion counterparts [81]. For reduction potentials, comparisons are made against electrochemical measurements [82].
High-Level Theory Benchmarking: NNPs are benchmarked against highly accurate computational methods like CCSD(T), considered the "gold standard" in quantum chemistry despite its computational expense [83]. This is particularly important for properties where experimental data is scarce.
Multi-Fidelity Transfer Learning: Recent approaches leverage transfer learning between different levels of theory, using abundant low-fidelity data (e.g., from DFT) to improve performance on sparse high-fidelity data (e.g., from CCSD(T) or experiments) [84]. Graph neural networks have shown particular promise in this area, demonstrating up to 8x performance improvements in low-data regimes [84].
Specialized Protocols for Charge-Related Properties
Specific experimental protocols have been developed for benchmarking charge-related properties:
Electron Affinity Calculation Protocol [81]:
- Optimize molecular geometries for both neutral and anionic states
- Perform single-point energy calculations on both systems
- Compute electron affinity as: EA = Eneutral - Eanion (converted to eV)
- Compare predictions against experimental gas-phase electron affinity measurements
- Analyze scaling behavior across molecular series of increasing size
Reduction Potential Benchmarking [82]:
- Evaluate NNPs on diverse molecular sets including main-group and organometallic species
- Compare NNP predictions against both experimental data and low-cost DFT methods
- Assess systematic trends across chemical space, particularly for organometallic species
Scaling Behavior Analysis [81]:
- Test NNPs on homologous series of increasing molecular size
- Analyze whether models capture correct physical scaling relationships
- Identify potential breakdown points for models trained primarily on small systems
The workflow below illustrates the complete benchmarking process for charge-related properties:
Figure 1: Benchmarking Workflow for NNP Validation. This diagram illustrates the comprehensive process for validating neural network potentials against both experimental data and high-level theoretical methods.
Key Benchmarking Studies and Quantitative Results
Electron Affinity Predictions Across Acene Series
A critical study examined how well NNPs predict electron affinities for linear acenes from naphthalene (2 rings) to undecacene (11 rings), testing both accuracy and scaling behavior [81]. The research compared multiple computational methods against available experimental data, with results summarized in the table below.
Table 1: Benchmarking Electron Affinity Predictions for Linear Acenes (values in eV) [81]
| Number of Rings | Experimental | GFN2-xTB | UMA-S | UMA-M | eSEN-S | ωB97M-V/def2-TZVPP |
|---|---|---|---|---|---|---|
| 2 (Naphthalene) | -0.19 | -0.195 | -0.428 | -0.387 | -0.374 | -0.457 |
| 3 (Anthracene) | 0.532 | 0.671 | 0.366 | 0.382 | 0.369 | 0.358 |
| 4 (Tetracene) | 1.04 | 1.233 | 0.890 | 0.925 | 0.958 | 0.930 |
| 5 (Pentacene) | 1.43 | 1.629 | 1.269 | 1.311 | 1.356 | 1.346 |
| 6 | - | 1.923 | 1.475 | 1.617 | 1.594 | 1.657 |
| 7 | - | 2.149 | 1.687 | 1.950 | 1.839 | 1.842 |
| 8 | - | 2.329 | 1.848 | 2.234 | 2.031 | 2.083 |
| 9 | - | 2.476 | 1.972 | 2.508 | 2.192 | 2.272 |
| 10 | - | 2.598 | 2.067 | 2.769 | 2.322 | 2.415 |
| 11 | - | 2.703 | 2.142 | 3.011 | 2.443 | 2.630 |
The benchmarking revealed that NNPs (UMA and eSEN models) performed comparably to DFT methods for electron affinity predictions across the acene series. Notably, these models correctly captured the physical scaling relationship where larger acenes exhibit higher electron affinities due to increased electron delocalization, despite lacking explicit Coulombic interaction terms in their architecture [81].
Reduction Potential and General Charge Property Validation
A comprehensive study evaluated OMol25-trained NNPs on experimental reduction potential and electron affinity data across diverse main-group and organometallic systems [82]. The benchmarking produced several key findings:
Table 2: Performance Summary of OMol25-Trained NNPs on Charge-Related Properties [82]
| Benchmark Category | System Types | NNP Performance | Comparison to Traditional Methods |
|---|---|---|---|
| Electron Affinity | Main-group molecules | Accurate prediction of gas-phase values | Comparable or superior to low-cost DFT and semiempirical methods |
| Reduction Potential | Organometallic species | High accuracy, outperforming expectations | Better performance than for main-group systems (reverse of DFT trend) |
| Charge Transfer | Diverse molecular pairs | Accurate coupling predictions | Neural networks outperform kernel ridge regression for electronic coupling [85] |
| Scaling Behavior | Large acenes (up to 30Å) | Correct physical scaling captured | Maintains accuracy beyond training set size limitations |
Surprisingly, the study found that OMol25-trained NNPs predicted charge-related properties of organometallic species more accurately than those of main-group species, contrary to trends observed with traditional DFT and semiempirical quantum mechanical methods [82]. This suggests that the NNPs are learning complex electronic relationships beyond simple physical approximations.
Implementing rigorous NNP benchmarks requires specific computational tools and resources. The following table details essential components for conducting charge-related property validation.
Table 3: Essential Research Reagents and Computational Tools for NNP Benchmarking
| Tool/Resource | Type | Function in Benchmarking | Key Features |
|---|---|---|---|
| OMol25 Dataset [15] | Dataset | Provides training data and benchmark structures | 100M+ calculations at ωB97M-V/def2-TZVPD level; diverse chemical space coverage |
| UMA Models [15] | Neural Network Architecture | Universal model for atoms across multiple datasets | Mixture of Linear Experts (MoLE) architecture; transfer learning capability |
| eSEN Models [15] | Neural Network Architecture | Conservative force predictions for molecular dynamics | Transformer-style architecture; equivariant spherical-harmonic representations |
| Rowan Platform [81] | Computational Infrastructure | Runs quantum calculations and NNP simulations | API access to NNPs and DFT methods; workflow automation |
| DP-GEN Framework [76] | Training System | Generates neural network potentials via active learning | Automated training data selection; transfer learning capability |
| Hirshfeld Charge Analysis [86] | Analytical Method | Partitions electron density for atomic charges | Measures charge density dissimilarities; reference for ML potentials |
Advanced Benchmarking Techniques and Architecture Considerations
Multi-Task Learning and Transfer Learning Strategies
Advanced benchmarking now incorporates sophisticated training strategies that impact model performance:
Multi-Task Electronic Hamiltonian Networks (MEHnet): A breakthrough approach from MIT researchers uses a single model to evaluate multiple electronic properties simultaneously, including dipole and quadrupole moments, electronic polarizability, and optical excitation gaps [83]. This multi-task framework demonstrates that property interdependencies can improve overall prediction accuracy compared to separate specialized models.
Transfer Learning in Multi-Fidelity Settings: Research demonstrates that graph neural networks can effectively leverage low-fidelity measurements (e.g., high-throughput screening data) to improve predictions on sparse, high-fidelity experimental data [84]. The benchmarking of these transfer learning approaches shows they can improve performance by up to 8x while using an order of magnitude less high-fidelity training data [84].
Two-Phase Training Schemes: The eSEN architecture employs an innovative two-phase training strategy where models are first trained for direct-force prediction, then fine-tuned for conservative force prediction [15]. This approach reduces training time by 40% while improving performance, representing a significant advancement in efficient model development.
Architectural Innovations and Their Benchmarking Implications
Recent architectural developments in NNPs have introduced critical capabilities for charge-related property prediction:
Equivariant Architectures: Models like eSEN and UMA utilize E(3)-equivariant networks that respect physical symmetries, ensuring property predictions remain consistent across rotational and translational transformations [15]. This is particularly important for charge transfer properties that depend on relative molecular orientations.
Mixture of Linear Experts (MoLE): The UMA architecture's MoLE framework enables effective knowledge transfer across datasets computed with different theoretical levels and basis sets [15]. Benchmarking shows this approach outperforms both naïve multi-task learning and single-task models, indicating genuine knowledge transfer occurs across chemical domains.
Attention Mechanisms and Adaptive Readouts: Graph neural networks with attention-based readout functions demonstrate significantly improved transfer learning capabilities compared to fixed aggregation functions [84]. These adaptive readouts are particularly valuable for molecular properties that depend on complex, non-local interactions like charge transfer.
The relationship between these architectural components and their benchmarking outcomes can be visualized as follows:
Figure 2: Architectural Features and Benchmarking Benefits. This diagram maps specific neural network potential architectural innovations to their demonstrated benefits in benchmarking studies.
Benchmarking neural network potentials on charge-related properties has revealed both remarkable capabilities and important limitations. The comprehensive validation studies conducted on OMol25-trained models demonstrate that modern NNPs can predict electron affinities, reduction potentials, and other charge-related properties with accuracy comparable to or exceeding traditional DFT methods, despite often lacking explicit physics-based treatment of long-range interactions [81] [82].
The most significant finding from recent benchmarking efforts is that NNPs exhibit surprisingly good transfer learning capabilities and scaling behavior, correctly capturing physical trends like increasing electron affinity with molecular size in acene systems [81]. However, challenges remain in ensuring these models maintain physical accuracy across the entire chemical space, particularly for very large systems where emergent inaccuracies may arise [81].
Future benchmarking efforts should focus on several key areas: (1) validating NNP performance on condensed-phase charge transfer processes, (2) establishing standardized benchmarking protocols for charge-related properties across diverse molecular classes, and (3) developing specialized architectures that explicitly incorporate physical constraints for charge interactions. As NNPs continue to evolve, rigorous benchmarking will remain essential for translating architectural advances into reliable chemical predictions, ultimately accelerating the discovery of new materials and medicines through computational means.
Benchmarking serves as the cornerstone of progress in computational chemistry, providing the rigorous, standardized framework necessary to transition from theoretical models to reliable scientific and decision-support tools. Within model validation research, benchmarking is the systematic process of evaluating and comparing the performance of computational methods against trusted reference data and established standards. This process is vital for assessing model accuracy, robustness, and practical utility across diverse chemical domains, from drug discovery to materials science. The proliferation of complex models, particularly machine-learned interatomic potentials (MLIPs) and AI-driven tools, has intensified the need for robust benchmarking. As noted in the introduction of MLIPAudit, "the field still lacks a standardised and comprehensive framework for evaluating MLIP performance" [7]. Community-driven leaderboards and standards emerge as a critical response to this challenge, transforming isolated validation efforts into a coordinated, transparent mechanism for tracking collective progress.
Current Benchmarking Platforms and Leaderboards
The computational chemistry community has developed several specialized platforms to address benchmarking needs across different sub-fields. These initiatives shift the evaluation focus from isolated error metrics to holistic performance assessments on downstream scientific tasks.
Table 1: Notable Benchmarking Platforms in Computational Chemistry
| Platform Name | Primary Focus | Key Metrics | Unique Features |
|---|---|---|---|
| MLIPAudit [7] | Machine-Learned Interatomic Potentials (MLIPs) | Stability, Transferability, Robustness, Accuracy on downstream tasks | Open, curated repository; supports diverse systems (proteins, molecular liquids); continuous leaderboard on HuggingFace |
| MLIP Arena [7] | MLIPs | Physical awareness, Stability, Reactivity, Predictive power | Leaderboard based on a compact, focused benchmark suite |
| Matbench Discovery [7] | Materials Science | Predictive accuracy for material properties | Easily extendable framework focused on materials discovery |
| Chemprop-MCP [87] | Chemical Property Prediction | Prediction accuracy on defined benchmarks (e.g., aqueous solubility) | Integration with AI workflows via Model Context Protocol (MCP) |
These platforms address a critical gap: standard energy and force validation errors, while necessary, are insufficient for estimating practical utility. As MLIPAudit notes, "models with very similar force validation error show significant variation in performance on a structural relaxation task" [7]. Leaderboards like these enforce a much-needed paradigm shift towards validation that reflects real-world simulation demands.
Community Standards and Methodological Guidelines
Beyond performance tracking, the establishment of community-sanctioned standards ensures methodological rigor and reproducibility. Prominent journals have begun formalizing these requirements, which function as a de facto benchmark for methodological quality.
Table 2: Key Community Standards from Drug Design, Development and Therapy Journal [88]
| Method Category | Practices Warranting Rejection | Required Best Practices |
|---|---|---|
| QSAR & ML Models | 2D-QSAR studies; Black-box models without interpretability/benchmarking | 3D+ models with rigorous validation; Curated training data; Independent test sets; Model interpretability |
| Docking & Virtual Screening | Reporting docking scores as absolute energies; Unvalidated hit lists | Full disclosure of preparation parameters; Benchmarking against known ligands; Experimental hit validation |
| Molecular Dynamics (MD) | Single, short trajectories; Poor system preparation; Over-interpretation | High-quality starting structures; Correct protonation states; Sufficient timescales; Multiple replicas |
| Free Energy Methods | Lack of error analysis/convergence checks; No experimental correlation | Rigorous sampling; Convergence analysis; Benchmarking against experimental data |
These standards address the "fundamental mismatch" between training data regimes and the demands of downstream applications [7]. They guard against the reporting of meaningless metrics and ensure that computational studies are biologically meaningful, transparent, and reproducible.
Quantitative Benchmarking Data and Performance Metrics
Effective benchmarking relies on quantitative, statistically rigorous comparisons. The field is moving beyond single-value error reporting towards approaches that quantify uncertainty and performance variability.
Table 3: Illustrative Benchmarking Results from Recent Studies
| Study & Method | System Benchmarked | Key Performance Metric | Result / Finding |
|---|---|---|---|
| GW Approximation vs. Coupled-Cluster [89] | 3d Transition-Metal Atoms & Molecules | Mean Absolute Error (MAE) for Ionization Potentials/Electron Attachment | G0W0@PBE0 MAE: 0.18-0.26 eV (atoms), 0.37-0.60 eV (molecules); More efficient than high-level wave function methods |
| Bayesian Hierarchical Models [90] | Saddle Search Algorithms (500 molecular systems) | Robustness, Computational Cost (energy/force calls) | Confirmed Conjugate Gradient (CG) offers higher overall robustness than L-BFGS; Supported design of adaptive workflows |
| TabPFN Foundation Model [91] | Small Tabular Datasets (<10,000 samples) | Classification/Regression Accuracy vs. Speed | Outperformed gradient-boosted decision trees with a 5,140x speedup in classification |
| IDA Vibrational Frequency Prediction [61] | Iminodiacetic Acid (IDA) | Prediction vs. Experimental Frequencies | All methods failed at high frequencies (>2200 cm⁻¹), highlighting that even established methods can fail. |
The application of advanced statistical models is itself a benchmark for rigor. As noted in the Bayesian hierarchical model study, this framework "rigorously quantifies performance metrics and their uncertainty, enabling a nuanced comparison of algorithmic strategies" rather than simple performance rankings [90].
Experimental Protocols for Benchmarking
A standardized benchmarking protocol is essential for generating fair and comparable results. The following workflow, implemented by frameworks like MLIPAudit, outlines a robust methodology for model evaluation.
Workflow Diagram Title: Model Benchmarking Protocol
Detailed Methodological Steps:
System Selection and Curation: Benchmarks must employ a diverse set of systems representative of real-world application domains. For MLIPs, this includes "small organic compounds, molecular liquids, proteins and flexible peptides" [7]. For quantum chemistry methods, select molecules with well-established reference data (e.g., coupled-cluster quality).
Reference Data Acquisition: Utilize trusted experimental data or high-fidelity computational results (e.g., ΔCCSD(T) for transition metals [89]). The quality of the benchmark is directly tied to the quality of its reference data.
Computational Configuration: Ensure consistent and reproducible software environments. Document all critical parameters: for quantum chemistry, this includes theory level, functional, and basis set [88] [61]; for MLIPs, this includes the software and model version.
Execution and Sampling: For dynamic properties, adequate sampling is critical. Molecular dynamics simulations require "sufficient timescales" and "multiple replicas" to ensure statistical robustness [88]. Avoid conclusions based on "single, short, or poorly prepared trajectories" [88].
Performance Metric Calculation: Move beyond basic error metrics. Calculate a suite of metrics relevant to the application, including stability, transferability, and accuracy on downstream tasks [7]. Employ statistical models to quantify uncertainty where possible [90].
Results Analysis and Reporting: Perform critical analysis to identify model failure modes and performance boundaries. As demonstrated in the IDA study, report clear limitations—such as the failure of all methods to predict high-frequency vibrations [61].
The Research Reagent Toolkit for Benchmarking
A standardized set of computational "reagents" is essential for conducting reproducible benchmarks. The table below details key resources and their functions in the validation process.
Table 4: Essential Research Reagents for Computational Benchmarking
| Tool / Resource | Function in Benchmarking | Examples / Standards |
|---|---|---|
| Reference Datasets | Provides ground truth for model validation | Aqueous solubility benchmarks [87]; ΔCCSD(T) data for transition metals [89] |
| Benchmarking Software | Automates evaluation pipelines and metric calculation | MLIPAudit suite [7]; Custom scripts for error analysis |
| Standardized Model Inputs | Ensures consistent starting points for comparisons | High-quality initial structures; Correct protonation/tautomer states [88] |
| Statistical Analysis Tools | Quantifies performance and uncertainty | Bayesian hierarchical models [90]; Standard regression metrics (MAE, RMSE) |
| Community Standards | Defines minimal acceptable methodological quality | Journal guidelines (e.g., rejection of unvalidated 2D-QSAR) [88] |
The Ecosystem of Progress: Standards, Tools, and Community
The synergistic relationship between community standards, benchmarking tools, and researcher engagement creates a powerful ecosystem that drives progress. This interconnected system ensures that model development is guided by real-world utility and rigorous validation.
Diagram Title: Benchmarking Ecosystem
This ecosystem functions as a continuous feedback loop. Community standards, such as those mandating experimental validation for virtual screening hits [88], set the baseline for quality. Benchmarking tools and leaderboards like MLIPAudit [7] provide the platform for transparent, comparative validation. Finally, the research community both utilizes these tools and contributes new models and data, which in turn reveals methodological gaps and drives the evolution of both standards and tools. The ultimate output is a trajectory of progress where models are not just academically interesting but are scientifically reliable and actionable.
Leaderboards and community standards are indispensable for tracking progress and ensuring the reliability of computational chemistry models. They collectively establish a framework for transparent, reproducible, and biologically meaningful research. The ongoing development of platforms like MLIPAudit and the enforcement of rigorous journal standards signify a maturation of the field. By adhering to these benchmarks and contributing to the ecosystem, researchers can accelerate the development of robust, trustworthy computational methods that genuinely advance scientific discovery and drug development.
Conclusion
Benchmarking is the cornerstone of reliable computational chemistry, providing the necessary validation to translate theoretical models into practical tools for drug discovery and biomedical research. A successful benchmarking strategy integrates rigorous statistical validation with an understanding of a model's applicability domain and its performance on real-world, downstream tasks. As the field advances, future efforts must focus on developing community-wide, blinded benchmarking challenges—similar to CASP in protein structure prediction—to foster transparency and continuous improvement. The adoption of more sophisticated benchmarks that probe dynamic behavior and complex biological interactions will be crucial for tackling unmet medical needs and accelerating the development of new therapeutics. By embracing these rigorous validation practices, researchers can confidently leverage computational power to drive the next generation of scientific breakthroughs.
References
- 1. Validation of computational results with experimental data [https://fiveable.me/computational-chemistry/unit-19/validation-computational-results-experimental-data/study-guide/pqxELTWdgS036XAN]
- 2. Verification and validation benchmarks [https://www.sciencedirect.com/science/article/abs/pii/S0029549307003548]
- 3. Verification, Validation and Uncertainty Quantification ... [https://www.asme.org/codes-standards/publications-information/verification-validation-uncertainty]
- 4. Activity prediction benchmarking [https://www.desertsci.com/2025/06/27/activity-prediction-benchmarking/]
- 5. Essential guidelines for computational method benchmarking [https://pmc.ncbi.nlm.nih.gov/articles/PMC6584985/]
- 6. A Record-Breaking Dataset to Train AI Models has Launched [https://newscenter.lbl.gov/2025/05/14/computational-chemistry-unlocked-a-record-breaking-dataset-to-train-ai-models-has-launched/]
- 7. MLIPAudit: A benchmarking tool for Machine Learned ... [https://arxiv.org/html/2511.20487v1]
- 8. Benchmarking OMol25-Trained Models on Experimental ... [https://rowansci.com/publications/omol25-redox]
- 9. Experimental validation of correlation peak universality in ... [https://www.sciencedirect.com/science/article/abs/pii/S0167732225004088]
- 10. LAMBench: A Benchmark for Large Atomistic Models [https://arxiv.org/html/2504.19578v2]
- 11. Extending quantum-mechanical benchmark accuracy to ... [https://www.nature.com/articles/s41467-025-63587-9]
- 12. A statistical framework for evaluating repeatability and ... [https://www.medrxiv.org/content/10.1101/2025.08.06.25333170v1.full-text]
- 13. High-Accuracy and High-Speed MOF Calculations with ... [https://matlantis.com/en/resources/blog/mofsimbench_matlantis/]
- 14. Comprehensive benchmarking of computational tools for ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00931-z]
- 15. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 16. Study of the Applicability Domain of the QSAR ... [https://www.mdpi.com/1420-3049/23/11/2756]
- 17. Efficiency of different measures for defining the applicability ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-017-0230-2]
- 18. Identifying domains of applicability of machine learning ... [https://www.nature.com/articles/s41467-020-17112-9]
- 19. reanalyzing a large-scale comparison of deep learning and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7292817/]
- 20. Uncertain of uncertainties? A comparison of uncertainty quantification... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00790-0]
- 21. FDA Announces Plan to Phase Out Animal Testing ... [https://www.fda.gov/news-events/press-announcements/fda-announces-plan-phase-out-animal-testing-requirement-monoclonal-antibodies-and-other-drugs]
- 22. New Approach Methodologies (NAMs) in Drug Development [https://www.thermofisher.com/blog/biotechnology/new-approach-methodologies-nams-in-drug-development/]
- 23. Regulatory acceptance of new approach methodologies ... [https://www.ema.europa.eu/en/human-regulatory-overview/research-development/ethical-use-animals-medicine-testing/regulatory-acceptance-new-approach-methodologies-nams-reduce-animal-use-testing]
- 24. Wiggle150: Benchmarking Density Functionals And Neural ... [https://chemrxiv.org/engage/chemrxiv/article-details/679284cc81d2151a02a622dd]
- 25. An Efficient Computational Chemistry Approach to ... [https://chemrxiv.org/engage/chemrxiv/article-details/68737a05728bf9025e3ae9c2]
- 26. A framework for evaluating the chemical knowledge and ... [https://www.nature.com/articles/s41557-025-01815-x]
- 27. Benchmarking Quantum Chemical Methods: Are We Heading ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5582598/]
- 28. Advancing data-driven chemistry by beating benchmarks [https://www.sciencedirect.com/science/article/abs/pii/S2589597422001265]
- 29. Better Benchmarking Improves Drug Development ... [https://www.intelligencia.ai/better-benchmarking-elevates-pharma-decision-making/]
- 30. Benchmarking R&D success rates of leading ... [https://www.sciencedirect.com/science/article/pii/S1359644625000042]
- 31. An Analysis Of Clinical Development Benchmarking ... [https://www.clinicalleader.com/doc/an-analysis-of-clinical-development-benchmarking-practices-0001]
- 32. Strategies for robust, accurate, and generalizable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607264/]
- 33. MEHC-Curation: A Python Framework for High-Quality ... [https://chemrxiv.org/engage/chemrxiv/article-details/69119f7cef936fb4a266439e]
- 34. Automated Workflows for Data Curation and Machine ... [https://pubmed.ncbi.nlm.nih.gov/39312162/]
- 35. Computational models for the assessment of manufactured ... [https://www.sciencedirect.com/science/article/pii/S2468111318300847]
- 36. Mlipaudit Benchmarks Machine Learned Interatomic ... [https://quantumzeitgeist.com/machine-mlipaudit-benchmarks-learned-interatomic-potentials-accurate-cost-effective/]
- 37. [2511.20487] MLIPAudit: A benchmarking tool for Machine ... [https://arxiv.org/abs/2511.20487]
- 38. A Standardized Benchmark for Machine-Learned ... [https://arxiv.org/html/2510.17187v1]
- 39. CatBench framework for benchmarking machine learning ... [https://www.sciencedirect.com/science/article/pii/S2666386425005673]
- 40. MP-ALOE: an r2SCAN dataset for universal machine ... [https://www.nature.com/articles/s41524-025-01834-9]
- 41. Benchmarking ML in ADMET predictions: the practical impact ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01041-0]
- 42. Revolutionizing pharmacology: AI-powered approaches in ... [https://www.sciencedirect.com/science/article/pii/S259009862500020X]
- 43. Development, validation and integration of (Q)SAR models to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6778835/]
- 44. mindrank-ai/PharmaBench [https://github.com/mindrank-ai/PharmaBench]
- 45. datagrok-ai/admetica [https://github.com/datagrok-ai/admetica]
- 46. ADMET Predictor® Modules [https://www.simulations-plus.com/software/admetpredictor/]
- 47. ADMET-AI: a machine learning ADMET platform for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11226862/]
- 48. Toxicity Prediction | QSTR | Adverse Drug Reactions [https://www.simulations-plus.com/software/admetpredictor/toxicity/]
- 49. A Review of Quantitative Structure–Activity Relationship ... [https://www.mdpi.com/2305-6304/13/9/799]
- 50. Benchmarking compound activity prediction for real-world ... [https://www.nature.com/articles/s42004-024-01204-4]
- 51. One size does not fit all: revising traditional paradigms for ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-00948-y]
- 52. Validation of OECD QSAR Toolbox profilers for ... [http://ui.adsabs.harvard.edu/abs/2025CmTox..3400356K/abstract]
- 53. AI-Integrated QSAR Modeling for Enhanced Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12525248/]
- 54. Quantitative Structure-Activity Relationship Models [https://www.emergentmind.com/topics/quantitative-structure-activity-relationship-qsar-models]
- 55. Resolving data bias improves generalization in binding ... [https://www.nature.com/articles/s42256-025-01124-5]
- 56. Recent advances in AI-driven protein-ligand interaction ... [https://www.sciencedirect.com/science/article/pii/S0959440X25000387]
- 57. TEMPL: A Template-Based Protein–Ligand Pose Prediction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570141/]
- 58. Data Leakage and Redundancy in the LIT-PCBA Benchmark [https://arxiv.org/html/2507.21404v1]
- 59. JARVIS-Leaderboard: a large scale benchmark of ... [https://www.nature.com/articles/s41524-024-01259-w]
- 60. Quality, Applicability, and Best Practices for Machine ... [https://www.sciencedirect.com/science/article/abs/pii/S2589597420303142]
- 61. Benchmarking computational chemistry approaches on ... [https://www.sciencedirect.com/science/article/abs/pii/S0019452223000183]
- 62. Random vs. Systematic Error | Definition & Examples [https://www.scribbr.com/methodology/random-vs-systematic-error/]
- 63. Random vs. Systematic Error [https://www.physics.umd.edu/courses/Phys276/Hill/Information/Notes/ErrorAnalysis.html]
- 64. A Step-by-Step Guide to Conducting Effective ... [https://www.comparables.ai/articles/step-by-step-guide-to-conducting-benchmarking-analysis]
- 65. Is there a set of updated, comprehensive benchmarks for ... [https://mattermodeling.stackexchange.com/questions/1354/is-there-a-set-of-updated-comprehensive-benchmarks-for-speed-comparison-between]
- 66. Step Forward Cross Validation for Bioactivity Prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC11245006/]
- 67. CACHE (Critical Assessment of Computational Hit-finding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9246350/]
- 68. Benchmark and Critical Evaluation for Machine Learning ... [https://ar5iv.labs.arxiv.org/html/2210.07237]
- 69. Discrepancies and error evaluation metrics for machine ... [https://www.nature.com/articles/s41524-023-01123-3]
- 70. A critical review on selecting performance evaluation ... [https://www.sciencedirect.com/science/article/pii/S2213343725043714]
- 71. Chemometric and computational modeling of ... [https://www.nature.com/articles/s41598-025-99823-x]
- 72. A Record-Breaking Dataset to Train AI Models has Launched [https://buildings.lbl.gov/news/computational-chemistry-unlocked-record-breaking-dataset-train-ai-models-has-launched]
- 73. From Simulation to Validation in Ensuring Quality and ... [https://www.mdpi.com/2076-3417/15/6/3107]
- 74. Benchmarking of Regression Algorithms on Major Evaluation ... [https://www.taylorfrancis.com/chapters/edit/10.1201/9781003606260-57/benchmarking-regression-algorithms-major-evaluation-criteria-stock-price-prediction-satish-kumar-singh-sheeba-praveen]
- 75. Comparative study between deep learning and QSAR ... [https://www.nature.com/articles/s41598-020-73681-1]
- 76. EMFF-2025: a general neural network potential for ... [https://www.nature.com/articles/s41524-025-01809-w]
- 77. Long-Range Interactions in High-Dimensional Neural ... [https://chemrxiv.org/engage/chemrxiv/article-details/688a8f0f728bf9025e2d8c6e]
- 78. Machine learning small molecule properties in drug ... [https://www.sciencedirect.com/science/article/pii/S2949747723000209]
- 79. ProQSAR: A Modular and Reproducible Framework for ... [https://chemrxiv.org/engage/chemrxiv/article-details/68e6a58abc2ac3a0e0f1aaed]
- 80. Comparative Study of (Q)SAR Models for Predicting the ... [https://www.sciencedirect.com/science/article/pii/S3050620425000491]
- 81. Studying Scaling in Electron-Affinity Predictions | Rowan [https://rowansci.com/blog/redox-scaling-study]
- 82. Benchmarking OMol25-Trained Models on Experimental ... [https://chemrxiv.org/engage/chemrxiv/article-details/68b75742728bf9025ede9923]
- 83. New computational chemistry techniques accelerate the ... [https://nse.mit.edu/computational-chemistry-predicting-molecules-materials/]
- 84. Transfer learning with graph neural networks for improved ... [https://www.nature.com/articles/s41467-024-45566-8]
- 85. Artificial neural networks for predicting charge transfer ... [https://pubmed.ncbi.nlm.nih.gov/33291923/]
- 86. Accuracy of Charge Densities in Electronic Structure ... [https://arxiv.org/html/2410.17866v1]
- 87. The Chemprop Model Context Protocol [https://chemrxiv.org/engage/chemrxiv/article-details/691b92f765a54c2d4a1e47b5]
- 88. Standards for Computational Methods in Drug Design and ... [https://www.dovepress.com/standards-for-computational-methods-in-drug-design-and-discovery-simpl-peer-reviewed-fulltext-article-DDDT]
- 89. Cluster Theory for 3d Transition Metals [https://chemrxiv.org/engage/chemrxiv/article-details/6814f462e561f77ed4c96183]
- 90. Bayesian Hierarchical Models for Quantitative Estimates ... [https://arxiv.org/abs/2505.13621]
- 91. Accurate predictions on small data with a tabular ... [https://www.nature.com/articles/s41586-024-08328-6]