Beyond the Training Set: Evaluating and Enhancing Out-of-Distribution Robustness in Molecular Property Predictors
The accurate prediction of molecular properties for compounds outside a model's training distribution is a critical frontier in AI-driven drug discovery.
Beyond the Training Set: Evaluating and Enhancing Out-of-Distribution Robustness in Molecular Property Predictors
Abstract
The accurate prediction of molecular properties for compounds outside a model's training distribution is a critical frontier in AI-driven drug discovery. This article provides a comprehensive analysis for researchers and drug development professionals, exploring the fundamental challenges of out-of-distribution (OOD) generalization. We systematically review the performance of state-of-the-art machine learning models, including graph neural networks and transformers, on established OOD benchmarks like BOOM. The content delves into innovative methodological strategies, from transductive learning and meta-learning to advanced uncertainty quantification, that aim to improve extrapolation. Finally, we present a rigorous framework for the validation and comparative analysis of molecular property predictors, underscoring the imperative of robust OOD evaluation for successful real-world application in biomedical research.
The OOD Generalization Challenge: Why Molecular Discovery is an Inherently Out-of-Distribution Problem
The discovery of high-performance materials and molecules often depends on identifying extremes—candidates with property values that fall outside the known distribution of existing data [1] [2]. Consequently, the ability to extrapolate to out-of-distribution (OOD) property values has become critical for both solid-state materials and molecular design [2]. In molecular contexts, "out-of-distribution" can refer to two distinct but sometimes overlapping concepts: extrapolation in the input space (unseen molecular structures, scaffolds, or chemical spaces) and extrapolation in the output space (unseen ranges of property values) [1] [2]. This distinction is crucial because models that perform well on one type of extrapolation may struggle with the other, leading to potentially misleading predictions in real-world drug discovery applications where both types of shifts commonly occur.
The practical implications of this challenge are significant. In critical applications such as drug screening or design, misleading estimations of molecular properties can result in tremendous waste of wet-lab resources and delay the discovery of novel therapies [3]. Molecular representation learning models typically assume that training and testing graphs come from identical distributions, but this closed-world assumption often breaks down when models are deployed in real-world scenarios [3] [4]. For example, a model trained on drugs inhibiting Gram-negative pathogens may perform poorly when screening for antibiotics against Gram-positive bacteria due to different pharmacological mechanisms [3].
Defining the OOD Spectrum in Molecular Science
Input Space Extrapolation: Navigating Structural Shifts
When OOD generalization is defined with respect to the input molecular space, extrapolation often involves generalization to unseen classes of molecular structures, scaffolds, or chemical environments [1] [2]. This includes scenarios such as training on artificial molecules and predicting natural products, or training on certain molecular scaffolds and predicting on entirely different scaffold classes [1]. In practice, this type of extrapolation frequently reduces to interpolation because test sets often remain within the same overall distribution as the training data in the representation space [1] [2]. This pattern is observed in predictive models using leave-one-cluster-out strategies and generative approaches aimed at generalizing to structures with varying atomic compositions or sizes [2].
Property Value Extrapolation: Targeting Performance Extremes
The second notion of extrapolation addresses the range of the predictive function—specifically, output material property values that may or may not correlate with extrapolation in the input materials space [2]. This work focuses on zero-shot extrapolation to property value ranges beyond the training distribution, which presents distinct challenges for classical machine learning models [1] [2]. When OOD generalization targets the range of predictive functions, traditional regression models face significant difficulties, leading some researchers to shift toward classification approaches for identifying OOD materials [1] [2].
Table: Comparison of OOD Types in Molecular Context
| Aspect | Input Space Extrapolation | Property Value Extrapolation |
|---|---|---|
| Definition | Generalization to unseen molecular structures/scaffolds | Generalization to unseen ranges of property values |
| Common Challenges | Often reduces to interpolation in representation space | Classical ML models struggle with regression extrapolation |
| Typical Approaches | Leave-one-cluster-out strategies, generative models | Classification of OOD materials, transductive methods |
| Practical Impact | Screening novel structural classes | Discovering high-performance extremes |
Methodological Frameworks for Molecular OOD Prediction
Bilinear Transduction for Property Value Extrapolation
Bilinear Transduction represents a transductive approach to OOD property prediction that reparameterizes the prediction problem [1] [2]. Rather than making property value predictions directly on new candidate materials, this method makes predictions based on a known training example and the difference in representation space between the two materials [1] [2]. During inference, property values are predicted similarly—based on a chosen training example and the difference between it and the new sample [2]. This approach enables extrapolation by learning how property values change as a function of material differences rather than predicting these values directly from new materials [1] [2].
The core innovation of this method lies in its ability to leverage analogical input-target relations in both training and test sets, enabling generalization beyond the training target support [1] [2]. Experimental results demonstrate that this approach improves extrapolative precision by 1.8× for materials and 1.5× for molecules, while boosting recall of high-performing candidates by up to 3× [2].
Prototypical Graph Reconstruction for Input Space Detection
For input space OOD detection in molecular graphs, the PGR-MOOD framework introduces a novel approach using diffusion model-based reconstruction [3]. This method addresses two significant challenges: (1) the inadequacy of Euclidean distance metrics for capturing complex graph structure similarities, and (2) the computational inefficiency of iterative denoising processes when applied to large molecular libraries [3].
PGR-MOOD operates by creating a series of prototypical graphs that align with in-distribution (ID) samples while distancing themselves from OOD ones [3]. During testing, it measures similarity between input molecules and these pre-constructed prototypical graphs using Fused Gromov-Wasserstein (FGW) distance, which comprehensively quantifies matching degree based on both discrete edges and continuous node features [3]. This approach eliminates the need to reconstruct every test graph, enabling scalable OOD detection for large molecular databases [3].
Consistent Semantic Representation Learning
The Consistent Semantic Representation Learning (CSRL) framework addresses challenges posed by activity cliffs and complex molecular entanglements that hinder accurate invariant substructure identification [4]. This approach explores the potential correlation between consistent semantic information across different molecular representation forms and molecular property prediction under distribution shifts [4].
CSRL comprises two key modules: a Semantic Uni-code (SUC) module that adjusts incorrect embeddings into correct embeddings across different molecular representation forms, and a Consistent Semantic Extractor (CSE) that leverages non-semantic information as training labels to guide the discriminator's learning [4]. This framework suppresses the model's reliance on non-semantic information in different molecular representation embeddings, enhancing OOD generalization capability [4].
Experimental Comparison of OOD Methodologies
Performance Evaluation on Molecular Benchmarks
Comprehensive evaluations across multiple molecular benchmarks reveal significant performance differences between OOD methodologies. On molecular graph datasets from MoleculeNet—including ESOL (aqueous solubility), FreeSolv (hydration free energies), Lipophilicity (distribution coefficients), and BACE (binding affinities)—transductive and reconstruction-based approaches demonstrate superior OOD detection capabilities compared to traditional methods [2] [3].
Table: OOD Detection Performance on Molecular Graphs (AUC Scores) [3]
| Method | ESOL | FreeSolv | Lipophilicity | BACE | Average |
|---|---|---|---|---|---|
| Random Forest | 0.742 | 0.768 | 0.715 | 0.731 | 0.739 |
| MLP | 0.751 | 0.781 | 0.724 | 0.748 | 0.751 |
| GCN | 0.793 | 0.812 | 0.768 | 0.792 | 0.791 |
| GIN | 0.811 | 0.834 | 0.785 | 0.816 | 0.812 |
| PGR-MOOD | 0.892 | 0.908 | 0.861 | 0.887 | 0.887 |
The PGR-MOOD framework demonstrates an average improvement of 8.54% in detection AUC and 8.15% in AUPR compared to baseline methods, accompanied by a 13.7% reduction in FPR95 (false positive rate at 95% true positive rate) [3]. These improvements come with substantially reduced computational costs in testing time and memory consumption, addressing critical constraints for large-scale molecular screening applications [3].
Property Value Extrapolation Performance
For property value extrapolation, Bilinear Transduction has been evaluated against established baselines including Ridge Regression, MODNet, and CrabNet across multiple materials and molecular datasets [1] [2]. The method consistently outperforms or performs comparably to baseline methods across diverse prediction tasks, with particularly strong performance in identifying top OOD candidates—the 30% of test samples with the highest property values [2].
Table: Extrapolative Precision on Molecular Property Prediction [2]
| Method | Molecular Datasets | Extrapolative Precision | OOD Recall |
|---|---|---|---|
| Ridge Regression | ESOL, FreeSolv, Lipophilicity, BACE | 0.18 | 1.0× |
| MODNet | ESOL, FreeSolv, Lipophilicity, BACE | 0.22 | 1.2× |
| CrabNet | ESOL, FreeSolv, Lipophilicity, BACE | 0.25 | 1.4× |
| Bilinear Transduction | ESOL, FreeSolv, Lipophilicity, BACE | 0.33 | 1.5× |
The Bilinear Transduction method improves extrapolative precision by 1.5× for molecules and boosts recall of high-performing candidates by up to 3× compared to non-transductive baselines [2]. This enhanced capability to identify true high-performance extremes while minimizing false positives significantly streamlines the virtual screening process in drug discovery pipelines [2].
Research Reagent Solutions: Computational Tools for OOD Molecular Prediction
Table: Essential Computational Tools for OOD Molecular Property Prediction
| Tool/Resource | Type | Function | Access |
|---|---|---|---|
| MatEx (Materials Extrapolation) | Software Library | Implements Bilinear Transduction for OOD property prediction | GitHub: learningmatter-mit/matex [2] |
| PGR-MOOD | Framework | Prototypical graph reconstruction for molecular OOD detection | Anonymous code: https://anonymous.4open.science/r/PGR-MOOD-53B3 [3] |
| DrugOOD | Benchmark Dataset | Curated molecular datasets with systematic OOD splits | Publicly available [4] |
| ADMEOOD | Benchmark Dataset | ADME property prediction with distribution shifts | Publicly available [4] |
| MoleculeNet | Benchmark Suite | Multiple molecular property prediction tasks | Publicly available [2] |
| CSRL Framework | Software Library | Consistent semantic representation learning for molecules | Details in publication [4] |
The evolving landscape of OOD molecular property prediction reveals a critical distinction between input space and property value extrapolation, each demanding specialized methodological approaches [1] [2] [3]. Transductive methods like Bilinear Transduction demonstrate significant advantages for property value extrapolation, while reconstruction-based approaches such as PGR-MOOD offer scalable solutions for input space OOD detection [2] [3]. The emerging paradigm of consistent semantic representation learning further addresses fundamental challenges posed by activity cliffs and molecular entanglement [4].
For researchers and drug development professionals, these advanced OOD detection and prediction capabilities enable more reliable virtual screening, reduce resource waste on false leads, and accelerate the discovery of novel molecular entities with extreme properties [2] [3]. As the field progresses, integrating these complementary approaches into unified frameworks promises to enhance the trustworthiness and real-world applicability of molecular property predictors across the drug discovery pipeline [5] [4].
The pursuit of novel therapeutics demands the discovery of materials and molecules with exceptional, often unprecedented, properties. By definition, these high-performing candidates possess property values that fall outside the distribution of known compounds, making the ability to extrapolate—to make accurate predictions on Out-of-Distribution (OOD) data—a cornerstone of accelerated drug discovery [2]. The failure of machine learning models to generalize in this context poses a significant bottleneck. Traditional models frequently experience a performance drop when encountering OOD samples and, more dangerously, can produce overconfident mispredictions, where the model assigns high confidence to an incorrect prediction [6]. Such errors are not merely statistical artifacts; they misdirect experimental resources, compromise virtual screening efforts, and can ultimately derail development pipelines, incurring substantial costs and delays. This guide objectively evaluates the OOD robustness of contemporary molecular property predictors, comparing their performance across key benchmarks to identify methodologies capable of navigating the challenging landscape of real-world drug discovery.
Quantitative Performance Comparison of OOD Prediction Methods
A critical evaluation of OOD performance requires examining models on standardized benchmarks where property values in the test set lie outside the range of the training data. The following tables summarize the extrapolative capabilities of leading methods against a transductive approach, Bilinear Transduction, on solid-state materials and molecules [2].
Table 1: OOD Prediction Performance on Solid-State Materials (Mean Absolute Error) [2]
| Dataset | Property | Ridge Regression | MODNet | CrabNet | Bilinear Transduction (Ours) |
|---|---|---|---|---|---|
| AFLOW | Bulk Modulus (GPa) | 74.0 ± 3.8 | 93.06 ± 3.7 | 59.25 ± 3.2 | 47.4 ± 3.4 |
| AFLOW | Debye Temperature (K) | 0.45 ± 0.03 | 0.62 ± 0.03 | 0.38 ± 0.02 | 0.31 ± 0.02 |
| AFLOW | Shear Modulus (GPa) | 0.69 ± 0.03 | 0.78 ± 0.04 | 0.55 ± 0.02 | 0.42 ± 0.02 |
| Matbench | Band Gap (eV) | 6.37 ± 0.28 | 3.26 ± 0.13 | 2.70 ± 0.13 | 2.54 ± 0.16 |
| Matbench | Yield Strength (MPa) | 972 ± 34 | 731 ± 82 | 740 ± 49 | 591 ± 62 |
| Materials Project | Bulk Modulus (GPa) | 151 ± 14 | 60.1 ± 3.9 | 57.8 ± 4.2 | 45.8 ± 3.9 |
Table 2: Extrapolative Precision for Identifying Top-Tier Candidates [2]
| System | Baseline Methods (Avg.) | Bilinear Transduction (Ours) | Precision Improvement |
|---|---|---|---|
| Solid-State Materials | - | - | 1.8x |
| Molecules | - | - | 1.5x |
Table 3: OOD Classification Performance [1]
| System | Metric | Baseline Methods | Bilinear Transduction (Ours) | Improvement |
|---|---|---|---|---|
| Materials | True Positive Rate (TPR) | - | - | 3.0x |
| Materials | Precision | - | - | 2.0x |
| Molecules | True Positive Rate (TPR) | - | - | 2.5x |
| Molecules | Precision | - | - | 1.5x |
The data demonstrates that Bilinear Transduction consistently achieves a lower Mean Absolute Error (MAE) on OOD predictions across a variety of material properties. More importantly for discovery applications, it significantly boosts extrapolative precision and the recall of high-performing OOD candidates, meaning a higher percentage of its predicted top candidates are truly top-tier, reducing the resources wasted on false leads [2] [1].
Detailed Experimental Protocols and Methodologies
Benchmarking OOD Property Prediction
Objective: To evaluate a model's zero-shot extrapolation capability, i.e., its ability to predict property values for samples that lie outside the range of the training data distribution [2]. Datasets: The protocol utilizes established benchmarks:
- Solids: AFLOW, Matbench, and the Materials Project (MP) datasets, covering properties like band gap, bulk/shear modulus, and Debye temperature [2].
- Molecules: Datasets from MoleculeNet, including ESOL (solubility), FreeSolv (hydration free energy), and Lipophilicity [2]. OOD Splitting: The held-out dataset is divided into an in-distribution (ID) validation set and an OOD test set of equal size. The OOD test set contains samples with property values strictly greater than the maximum value seen in the training set to focus evaluation on pure extrapolation [2]. Evaluation Metrics:
- OOD Mean Absolute Error (MAE): Measures prediction accuracy on the OOD test set [2].
- Extrapolative Precision: The fraction of true top OOD candidates (e.g., top 30% by property value in the entire held-out set) correctly identified among the model's top predicted candidates [2].
- Recall: The proportion of actual top OOD candidates successfully retrieved by the model [2].
Bilinear Transduction Workflow
The core innovation of this approach is a reparameterization of the prediction problem to facilitate extrapolation [2] [1].
- Input Representation: Materials (e.g., compositions) or molecules (e.g., graphs) are converted into a fixed-length vector representation.
- Bilinear Model: Instead of predicting a property value ( y ) for a new test material ( xt ) directly, the model learns to predict the value based on a known training example ( xs ) and their difference in representation space.
- Inference: For a test sample ( xt ), a training sample ( xs ) is selected (e.g., via similarity), and the property value is predicted as ( \hat{yt} = ys + f(xs, xt - x_s) ), where ( f ) is the learned bilinear function. This allows the model to learn how property values change as a function of material differences, which is more amenable to extrapolation than predicting absolute values from new materials [2] [1].
Evaluating and Mitigating Overconfident Errors
Objective: To assess and improve a model's uncertainty estimation, particularly for OOD samples, to reduce overconfident incorrect predictions [6]. Protocol:
- Model Modification: Replace the standard Softmax output layer in a classifier with a normalizing flow-based density estimator, as in the Posterior Network (AttFpPost). This enhances the model's ability to distinguish between in-distribution and out-of-distribution data [6].
- Evaluation Scenarios: The model is tested on:
- Synthetic OOD Data: To simulate domain shift.
- ADMET Prediction Tasks: Critical tasks in drug development (Absorption, Distribution, Metabolism, Excretion, and Toxicity).
- Ligand-Based Virtual Screening (LBVS): Assessing early enrichment capability [6]. Outcome: Models equipped with improved uncertainty quantification (like AttFpPost) demonstrate a marked reduction in overconfident errors on OOD samples compared to vanilla models using Softmax [6].
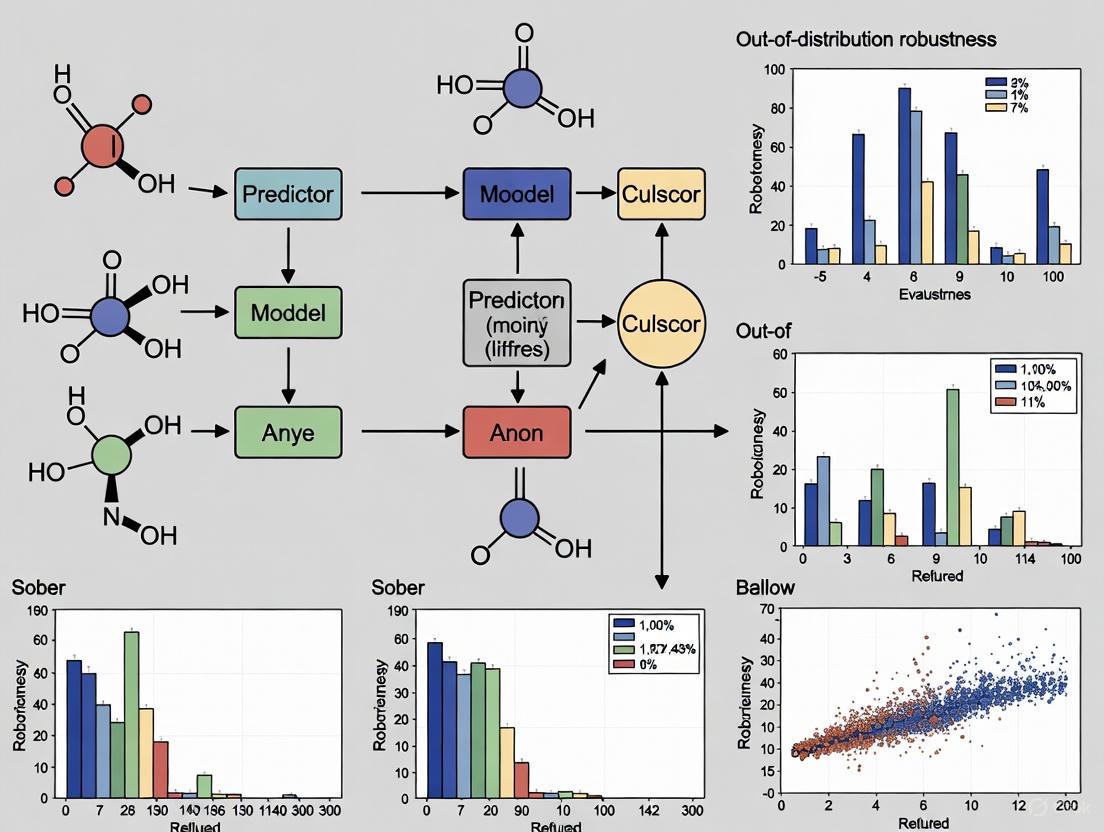
Visualizing the OOD Generalization Challenge and Solutions
The following diagrams illustrate the core problem of OOD generalization in drug discovery and the logical workflow of a robust evaluation protocol.
The OOD Generalization Gap in Drug Discovery
Protocol for Robust OOD Model Evaluation
The Scientist's Toolkit: Key Research Reagents and Solutions
This section details essential computational tools and datasets used in the featured experiments for benchmarking OOD robustness.
Table 4: Essential Research Toolkit for OOD Robustness Evaluation
| Item Name | Type | Function/Benefit | Source/Implementation |
|---|---|---|---|
| Bilinear Transduction | Algorithm | Enables extrapolation by learning property changes as a function of input differences. | GitHub: learningmatter-mit/matex [2] |
| AttFpPost (Posterior Network) | Model Architecture | Reduces overconfident errors on OOD samples via normalizing flows for better uncertainty estimation. | Citation: Patterns Journal [6] |
| AFLOW, Matbench, Materials Project | Data Benchmarks | Curated datasets for solid-state materials property prediction, enabling standardized OOD testing. | AFLOW API; Matbench [2] |
| MoleculeNet | Data Benchmarks | A collection of molecular property datasets (ESOL, FreeSolv, etc.) for benchmarking OOD generalization in molecules. | MoleculeNet [2] |
| GUEST Toolbox | Software Tool | A Python tool for the fair design and benchmarking of Drug-Target Interaction (DTI) prediction models, addressing data leakage. | GitHub: ML4BM-Lab/GraphEmb [7] |
| CleverHans & Foolbox | Software Library | Frameworks for generating adversarial examples to test and enhance model robustness against malicious inputs. | CleverHans GitHub; Foolbox Docs [8] |
The quantitative data and experimental protocols presented in this guide underscore a critical finding: traditional machine learning models exhibit significant vulnerabilities when predicting Out-of-Distribution properties, leading to overconfident errors that directly impede the drug discovery process. The evaluation of methods like Bilinear Transduction and uncertainty-aware models such as AttFpPost demonstrates that algorithmic choices which explicitly account for OOD generalization—through transduction or enhanced uncertainty quantification—can deliver substantially improved extrapolative precision and recall. For researchers and development professionals, this implies that the selection of a molecular property predictor must be guided not only by its in-distribution accuracy but, more importantly, by its rigorously tested OOD robustness. Integrating these robust methodologies and the accompanying toolkit into discovery pipelines is no longer optional but essential for efficiently identifying genuine, high-performance candidates and building a more trustworthy AI-driven future for pharmaceuticals.
The application of machine learning (ML) in molecular and materials discovery represents a paradigm shift in scientific research. However, a critical challenge undermines its real-world utility: models often fail to make accurate predictions on out-of-distribution (OOD) data. Molecular discovery is inherently an OOD prediction problem; discovering novel molecules that extend the boundaries of known chemistry requires models that can generalize to regions of chemical space beyond the training distribution [9]. Despite the importance of OOD performance, traditional benchmarks have predominantly evaluated models on in-distribution (ID) data, where test sets are randomly drawn from the same distribution as training data. This approach has led to overly optimistic performance assessments and models ill-equipped for practical discovery tasks [10].
This guide examines emerging benchmarks specifically designed for evaluating OOD generalization in molecular and materials property prediction. We focus on the recently introduced BOOM (Benchmarks for Out-Of-distribution Molecular property predictions) framework alongside other complementary initiatives [9] [11] [12]. By comparing their methodologies, experimental protocols, and key findings, we provide researchers with a comprehensive understanding of the current landscape and performance gaps in OOD prediction.
Benchmark Framework Comparison
The pressing need for systematic OOD evaluation has spurred the development of several benchmarking frameworks across domains. These frameworks employ different strategies to create meaningful distribution shifts between training and test data.
Table 1: Overview of OOD Benchmarking Frameworks
| Framework | Domain | OOD Splitting Strategy | Core Evaluation Focus | Key Contribution |
|---|---|---|---|---|
| BOOM [9] [12] | Molecular Property Prediction | Property-value based (tail-end of distribution) | Extrapolation to extreme property values | First large-scale benchmark for OOD molecular property prediction |
| Structure-based OOD Materials Benchmark [10] | Materials Property Prediction | Structure-based clustering (5 methods) | Generalization to novel material structures | Comprehensive benchmark for inorganic materials using structure-based GNNs |
| ImageNet-X/FS-X [13] [14] | Computer Vision | Semantic & covariate shifts | Detection under challenging real-world shifts | Benchmark for vision-language models with progressive difficulty |
| OpenMIBOOD [15] | Medical Imaging | Covariate-shifted ID, near-OOD, far-OOD | OOD detection in medical contexts | Domain-specific benchmark for healthcare AI reliability |
| MatEx (Bilinear Transduction) [2] | Molecules & Materials | Property-value based (zero-shot extrapolation) | Transductive extrapolation to high-value candidates | Novel method improving recall of high-performing OOD candidates |
BOOM: A Deep Dive into Molecular OOD Benchmarking
Experimental Design and Methodology
BOOM addresses a significant gap in molecular ML by providing the first standardized benchmark for assessing OOD generalization in molecular property prediction. Its methodology is built around several key design choices:
Property-based OOD Splitting: Unlike input-based splitting strategies, BOOM defines OOD with respect to model outputs, creating test sets from molecules with property values at the tail ends of the distribution. This directly aligns with molecule discovery goals where researchers seek materials with exceptional properties [9].
Dataset Composition: BOOM incorporates 10 molecular property datasets: 8 from QM9 (including isotropic polarizability, HOMO-LUMO gap, and dipole moment) and 2 from the 10k Dataset (density and solid heat of formation) [9].
Splitting Protocol: For each property, BOOM fits a kernel density estimator to the property values and selects molecules with the lowest probabilities (lowest 10% for QM9, lowest 1000 molecules for 10k Dataset) for the OOD test set. The remaining molecules are used for training and ID testing with random sampling [9].
Model Coverage: The benchmark evaluates over 140 combinations of models and tasks, including traditional ML (Random Forest with RDKit features), graph neural networks (GNNs) like Chemprop and TGNN, and transformer-based models (ChemBERTa, MolFormer) [9] [12].
The following diagram illustrates BOOM's experimental workflow from dataset preparation through to performance evaluation:
Key Experimental Findings from BOOM
BOOM's comprehensive evaluation reveals significant challenges in OOD generalization:
No Universal Performer: No single model achieved strong OOD generalization across all tasks. Even the top-performing model exhibited an average OOD error 3× larger than its in-distribution error [9] [12].
Inductive Bias Advantage: Deep learning models with high inductive bias (particularly certain GNN architectures) performed well on OOD tasks with simple, specific properties, suggesting that architectural choices should align with property characteristics [9].
Foundation Model Limitations: Current chemical foundation models with transfer and in-context learning showed promise for data-limited scenarios but did not demonstrate strong OOD extrapolation capabilities, indicating room for improvement in pretraining strategies [9].
Representation Impact: Molecular representation (SMILES, graphs, descriptors) significantly influenced OOD performance, with different excelling in different property prediction tasks [9].
Complementary OOD Benchmarks in Materials Science
Structure-based Materials Benchmark
A 2024 benchmark study focused on structure-based graph neural networks for inorganic materials property prediction proposed five distinct categories of OOD test sets based on crystal structure clustering [10]. This approach addresses the limitation of composition-based descriptors by incorporating structure-based representations like Orbital Field Matrix (OFM) for clustering.
Key findings from this benchmark include:
- Performance Overestimation: State-of-the-art GNN models that top leaderboards in conventional benchmarks (e.g., coGN, coNGN) showed significant performance drops on OOD test sets, demonstrating that reported superior performances were overestimated due to dataset redundancy [10].
- Generalization Gap: All algorithms performed worse on OOD tasks compared to their baseline MatBench performance, with an average performance drop that highlights a crucial generalization gap in realistic material prediction [10].
- Robust Performers: CGCNN, ALIGNN, and DeeperGATGNN demonstrated more robust OOD performance compared to current top MatBench models, providing insights for architectural improvements [10].
Transductive Approaches for OOD Extrapolation
The MatEx framework introduces a different approach to OOD property prediction using Bilinear Transduction, which reformulates the prediction problem by learning how property values change as a function of material differences rather than predicting values directly from new materials [2].
Table 2: Performance Comparison of OOD Methods on Solid-State Materials
| Method | Bulk Modulus MAE | Debye Temperature MAE | Shear Modulus MAE | Extrapolative Precision | OOD Recall |
|---|---|---|---|---|---|
| Bilinear Transduction [2] | Lower than baselines | Lower than baselines | Lower than baselines | 1.8× improvement | 3× boost |
| Ridge Regression [2] | Higher | Higher | Higher | Baseline | Baseline |
| MODNet [2] | Higher | Higher | Higher | Lower | Lower |
| CrabNet [2] | Higher | Higher | Higher | Lower | Lower |
This method demonstrated significant improvements in extrapolative precision (1.8× for materials, 1.5× for molecules) and boosted recall of high-performing candidates by up to 3× compared to conventional approaches [2].
Experimental Protocols and Methodologies
OOD Splitting Strategies
Different benchmarks employ distinct strategies for creating meaningful train-test splits:
- Property-based Splitting (BOOM): Uses kernel density estimation on property values to identify tail-end samples for OOD testing [9].
- Structure-based Clustering: Employs structural descriptors and clustering algorithms to identify novel material structures absent from training [10].
- Scaffold-based Splitting: Groups molecules by their Bemis-Murcko scaffolds and assigns entire scaffolds to different splits, testing generalization to novel molecular frameworks [16].
- Similarity-based Splitting: Uses chemical similarity clustering (K-means on ECFP4 fingerprints) to create challenging OOD splits where test molecules are distant from training examples in chemical space [16].
Evaluation Metrics
Comprehensive OOD evaluation requires multiple metrics:
- Performance Degradation: Comparison of ID vs. OOD performance (e.g., MAE ratio) [9] [10].
- Extrapolative Precision: Fraction of true top OOD candidates correctly identified among predicted top candidates [2].
- OOD Recall: Ability to retrieve high-performing OOD candidates [2].
- Ranking Consistency: Correlation between ID and OOD performance rankings across models [16].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools for OOD Molecular Property Prediction
| Tool/Resource | Type | Function | Relevance to OOD Evaluation |
|---|---|---|---|
| QM9 Dataset [9] | Dataset | 133,886 small organic molecules with quantum chemical properties | Primary benchmark dataset for molecular OOD evaluation |
| RDKit [9] [2] | Software | Cheminformatics and molecular descriptor generation | Featurization for traditional ML models and fingerprint generation |
| Graph Neural Networks [9] [10] | Model Architecture | Message-passing networks on molecular graphs | State-of-the-art for structure-property relationship learning |
| SMILES [9] [2] | Representation | String-based molecular representation | Input for transformer-based models and language approaches |
| Kernel Density Estimation [9] | Statistical Method | Probability density function estimation | Identifying low-probability samples for OOD test set creation |
| Bilinear Transduction [2] | Algorithm | Transductive extrapolation method | Improving recall of high-performing OOD candidates |
Performance Insights and Research Implications
The collective findings from these benchmarks reveal several critical patterns:
ID Performance ≠ OOD Performance: Strong in-distribution performance does not guarantee out-of-distribution generalization. The correlation between ID and OOD performance varies significantly based on the splitting strategy, with scaffold splitting showing stronger correlation (Pearson r ∼ 0.9) than cluster-based splitting (r ∼ 0.4) [16].
Architecture Matters: Model architecture significantly impacts OOD robustness. GNNs with strong inductive biases often outperform more flexible transformer architectures on OOD tasks, particularly for properties with clear structure-property relationships [9] [10].
Data Generation Impact: How OOD data is generated substantially influences benchmark difficulty. Cluster-based splitting using chemical similarity poses the hardest challenge for both classical ML and GNN models [16].
Domain-Specific Challenges: OOD detection methods that perform well in computer vision domains do not necessarily translate to scientific applications, underscoring the need for domain-specific benchmarks [15].
The following diagram summarizes the relationship between different OOD splitting strategies and their impact on model generalization:
The development of specialized benchmarks like BOOM represents a crucial step toward more reliable and deployable molecular machine learning models. The consistent finding across all benchmarks—that current state-of-the-art models struggle with OOD generalization—highlights a fundamental challenge in the field.
Moving forward, researchers should:
- Prioritize OOD performance alongside ID metrics when developing new models
- Consider multiple OOD splitting strategies to comprehensively assess generalization
- Explore architectural innovations that explicitly incorporate inductive biases for molecular systems
- Develop transductive methods and transfer learning strategies specifically designed for OOD scenarios
As the field progresses, these OOD benchmarks will play an increasingly vital role in guiding the development of molecular property predictors that can truly accelerate scientific discovery by reliably identifying novel materials with exceptional properties.
The pursuit of reliable machine learning (ML) models for molecular property prediction represents a cornerstone of modern computational chemistry and drug discovery. These models promise to accelerate the identification of novel molecules with desirable properties, from pharmaceutical compounds to sustainable energy materials. However, their real-world utility hinges on a critical factor: robustness to Out-of-Distribution (OOD) data. Molecular discovery is, by its very nature, an OOD problem; the goal is to identify molecules that extend beyond the boundaries of known chemical space or exhibit properties that extrapolate beyond the training data [9]. A model that performs excellently on in-distribution (ID) data but fails on OOD data offers limited practical value, potentially misguiding discovery campaigns.
Recent large-scale benchmarking studies have provided stark, quantitative evidence of a significant performance gap between ID and OOD settings. This guide synthesizes the latest evidence on this gap, compares the OOD performance of various molecular property prediction models, and details the experimental methodologies and emerging solutions aimed at building more robust ML systems for science.
Empirical Evidence: Documenting the Performance Gap
Systematic evaluations reveal that OOD generalization remains a formidable challenge for state-of-the-art models. The BOOM (Benchmarking Out-Of-distribution Molecular property predictions) study, a comprehensive analysis of over 140 model-and-task combinations, found that even the top-performing models exhibited an average OOD error that was 3x larger than their in-distribution error [9]. This finding is pivotal, as it demonstrates that the gap is not a minor inconvenience but a substantial degradation in model performance.
Table: Summary of OOD Performance Gaps from Key Studies
| Study / Benchmark | Key Finding on OOD Performance | Context / Models Evaluated |
|---|---|---|
| BOOM Benchmark [9] | Top-performing model's average OOD error was 3x larger than its ID error. | Evaluation of 12+ ML models across 10 molecular property prediction tasks. |
| ACS for Multi-Task GNNs [17] | Adaptive Checkpointing with Specialization (ACS) outperformed standard MTL by up to 10.8% and single-task learning by 15.3% on ClinTox, mitigating negative transfer. | Multi-task Graph Neural Networks on MoleculeNet benchmarks (ClinTox, SIDER, Tox21). |
| OOD Detection Survey [18] | ML models are vulnerable to distribution shifts; performance can be severely impacted by covariate and concept shifts. | Broad survey of distribution shift handling methods in machine learning. |
| Molecular Property Prediction Review [16] | The correlation between ID and OOD performance is highly dependent on the data splitting strategy, weakening significantly under challenging splits. | Evaluation of 12 models, including Random Forests and GNNs, across 8 datasets with 7 splitting strategies. |
The performance drop is not uniform across all OOD scenarios. The relationship between ID and OOD performance is strongly influenced by how the OOD data is generated. For instance, while a strong positive correlation (Pearson r ~ 0.9) between ID and OOD performance exists under simple scaffold splits, this correlation weakens significantly (Pearson r ~ 0.4) under more challenging, cluster-based data splits [16]. This indicates that model selection based solely on ID performance is an unreliable strategy for applications requiring OOD robustness.
Experimental Protocols: How the OOD Gap is Measured
Benchmark Creation and OOD Splitting Strategies
A critical step in quantifying OOD performance is the methodology used to split data into in-distribution and out-of-distribution sets. The following workflows and strategies are central to current research.
Diagram: Workflow for Benchmarking OOD Generalization
The diagram above outlines the general workflow for creating an OOD benchmark. The specific splitting strategies are crucial and include:
- Property-Based Splitting (Output Space): This method, used in the BOOM benchmark, defines OOD with respect to the model's prediction target. A kernel density estimator is fitted to the distribution of a molecular property's values. Molecules with the lowest probability densities—those at the tail ends of the distribution—are assigned to the OOD test set. This directly tests a model's ability to extrapolate to novel property values, which is central to molecule discovery [9].
- Scaffold-Based Splitting (Input Space): This approach groups molecules based on their Bemis-Murcko scaffolds (the core molecular framework). The test set contains molecules with scaffolds that are absent from the training set. This evaluates a model's ability to generalize to novel chemical structures [17] [16].
- Cluster-Based Splitting (Input Space): Molecules are clustered using their chemical fingerprints (e.g., ECFP4) and a clustering algorithm like K-means. Entire clusters are held out for the test set, creating a more challenging OOD scenario that often poses the hardest generalization challenge for models [16].
Model Training and Evaluation
Once splits are established, models are trained exclusively on the training set. Their performance is then evaluated separately on the ID test set (drawn from the same distribution as the training data) and the OOD test set. The key metrics, such as Mean Absolute Error (MAE) for regression or Area Under the Curve (AUC) for classification, are compared directly to calculate the performance gap [9] [16].
Comparative Analysis of Model Performance
The BOOM benchmark provides a broad overview of how different model classes handle OOD data. The evaluation included traditional machine learning models, Graph Neural Networks (GNNs) with various inductive biases, and transformer-based chemical foundation models.
Table: OOD Performance of Model Classes on Molecular Property Prediction
| Model Class | Example Models | Key OOD Findings | Notable Strengths & Weaknesses |
|---|---|---|---|
| Traditional ML | Random Forest (with RDKit features) | Struggles with challenging OOD splits like cluster-based. | Simple, but relies heavily on the quality and completeness of hand-crafted molecular descriptors. |
| Graph Neural Networks (GNNs) | Chemprop, TGNN, IGNN, EGNN, MACE | Performance varies with architecture and inductive bias. Models with high inductive bias can perform well on OOD tasks with simple, specific properties [9]. | Strong permutational invariance. E(3)-invariant/equivariant models can better capture geometric physics. |
| Transformers / Foundation Models | MolFormer, ChemBERTa, Regression Transformer, ModernBERT | Current chemical foundation models do not show strong OOD extrapolation capabilities consistently across tasks [9]. | Promising for limited data via transfer learning, but pretraining on large corpora does not guarantee OOD robustness. |
| Specialized GNN Architectures | D-MPNN, ACS (Multi-task GNN) | Can match or surpass performance of other models; ACS effectively mitigates negative transfer in imbalanced data [17]. | Architectural choices like directed messaging (D-MPNN) or adaptive checkpointing (ACS) can enhance robustness. |
A critical finding is that no single existing model achieves strong OOD generalization across all diverse tasks [9]. This underscores OOD property prediction as a "frontier challenge" for the field. Furthermore, the assumption that large foundation models will automatically solve this problem is not yet supported by evidence; their pretraining on vast chemical datasets does not necessarily confer robust OOD extrapolation capabilities [9].
Mitigation Strategies: Bridging the OOD Gap
Several advanced techniques have been developed to specifically address and reduce the OOD performance gap.
Diagram: The ACS Method for Mitigating Negative Transfer
- Adaptive Checkpointing with Specialization (ACS): Designed for multi-task GNNs, ACS combats negative transfer—where updates from one task degrade performance on another. It uses a shared backbone for general representation learning but employs task-specific heads. Crucially, it checkpoints the best backbone-head pair for each task whenever a new minimum validation loss is achieved for that task. This approach allows beneficial parameter sharing while protecting individual tasks from detrimental interference, significantly improving performance in low-data and imbalanced regimes [17].
- Confidence Optimal Transport (COT/COTT): This method addresses the underestimation of OOD error by leveraging optimal transport theory to provide more robust estimates of model performance on OOD data without labels. It is particularly effective in the presence of pseudo-label shift (discrepancy between predicted and true OOD label distributions). An empirically-motivated variant, COTT, further improves accuracy by applying thresholding to individual transport costs [19].
- Architectural Inductive Biases: Incorporating physical and chemical priors into model architectures can enhance OOD generalization. For instance, E(3)-equivariant GNNs, which respect the symmetries of 3D space, can better leverage geometric information, potentially leading to more robust predictions on unseen molecular structures [9].
Table: Essential Research Reagents for OOD Molecular Property Prediction
| Resource Name | Type | Primary Function in OOD Research |
|---|---|---|
| BOOM Benchmark [9] | Benchmark Suite | Standardized benchmark for assessing OOD generalization performance across 10 molecular property datasets. |
| QM9 Dataset [9] | Molecular Dataset | A well-known dataset of 133,886 small organic molecules with quantum mechanical properties, used for training and evaluation. |
| MoleculeNet [17] | Benchmark Suite | A collection of molecular datasets for benchmarking ML models, often used with scaffold splitting for OOD evaluation. |
| RDKit [9] | Cheminformatics Library | Used to generate molecular descriptors, fingerprints, and scaffolds for featurization and data splitting. |
| Graph Neural Networks (GNNs) | Model Architecture | Learns directly from molecular graph structure, providing a strong inductive bias for molecular data. |
| ACS Training Scheme [17] | Algorithm/Method | A training scheme for multi-task GNNs that mitigates negative transfer, enabling accurate prediction with as few as 29 labeled samples. |
| COT/COTT Algorithm [19] | Algorithm/Method | Provides robust estimates of model performance on OOD data without requiring labeled OOD examples. |
The evidence is clear and consistent: a significant performance gap, quantified as a 3x increase in error, exists between in-distribution and out-of-distribution settings for molecular property predictors. This gap poses a substantial risk to the reliability of AI-driven discovery pipelines. Addressing this challenge requires a multi-faceted approach: using rigorous benchmarking practices like those in BOOM, adopting advanced mitigation strategies like ACS and COT, and developing models with stronger physical and chemical inductive biases. For researchers and professionals in drug development, moving beyond in-distribution metrics and proactively evaluating OOD robustness is no longer optional but essential for building trustworthy and effective predictive models.
Architectural and Algorithmic Innovations for Improved OOD Extrapolation
The discovery of high-performance materials and molecules fundamentally depends on identifying extremes with property values that fall outside known distributions [2] [1]. Traditional machine learning models excel at interpolation within their training data but face significant challenges when making predictions for out-of-distribution (OOD) property values, a critical capability for accelerating scientific discovery [2]. This limitation is particularly problematic in virtual screening workflows, where the objective is to identify high-performing OOD candidates from known compounds with unknown properties [2] [1]. Transductive learning approaches, particularly Bilinear Transduction, have emerged as a promising framework for addressing this fundamental challenge in molecular and materials informatics.
The core problem stems from how conventional machine learning models generalize. Classical supervised learning typically struggles with extrapolating property predictions through regression when test samples fall outside the training distribution [1]. Consequently, many approaches have shifted toward classifying OOD materials instead of performing direct regression [1]. Bilinear Transduction represents a paradigm shift in this landscape by reformulating the prediction problem itself, moving from absolute property prediction to relative difference estimation, enabling more accurate zero-shot extrapolation to unprecedented property ranges [2] [1].
Understanding Transductive Learning: A Conceptual Framework
Inductive Versus Transductive Learning Paradigms
In machine learning, a critical distinction exists between inductive and transductive learning approaches [20]. Inductive learning follows the traditional supervised pattern: reasoning from observed training cases to general rules, which are then applied to test cases [20]. This approach builds a predictive model from seen data samples in the form of weights that can be applied to unseen samples [7]. Most conventional machine learning models used in materials informatics operate under this paradigm.
In contrast, transductive learning represents a different reasoning approach: moving from observed, specific training cases to specific test cases without intermediary general rules [20]. Transductive methods do not build a predictive model with weights that can be applied to unseen samples [7]. Instead, they use all available data—both training and test instances—to generate predictions directly. This fundamental difference in approach enables transductive methods to leverage relationships between test samples and training data more effectively, particularly valuable when dealing with distribution shifts [7].
The Problem of Data Leakage in Transductive Evaluation
A significant challenge in evaluating transductive methodologies lies in preventing data leakage during feature generation [7]. When improperly implemented, transductive approaches can exhibit artificially inflated performance metrics because information from the test set may inadvertently influence feature creation [7]. This has been particularly observed in drug-target interaction prediction, where transductive models have demonstrated near-optimal performance due to evaluation artifacts rather than genuine predictive capability [7]. Proper benchmarking requires careful experimental design to ensure fair comparison between inductive and transductive approaches, often involving specific dataset splitting strategies that isolate test information during training [7].
Bilinear Transduction: Core Methodology and Implementation
Theoretical Foundation and Mathematical Reformulation
Bilinear Transduction addresses the OOD prediction problem through a fundamental reparameterization of the prediction task [1] [21]. Rather than making property value predictions directly from a new candidate material's features, predictions derive from a known training example and the difference in representation space between the two materials [1]. This approach enables extrapolation by learning how property values change as a function of material differences rather than predicting these values from new materials in isolation [2].
The core innovation lies in decomposing the input variable into an anchor (a variable in the input space) and a delta (the difference between the input variable and the anchor) [21]. During inference, property values are predicted based on a chosen training example and the difference between it and the new sample [1]. This transformation effectively converts an out-of-support learning problem into an out-of-combination problem, which can be more tractable if the reparameterized training and test data distributions satisfy certain assumptions [21].
Implementation Workflow
The following diagram illustrates the conceptual workflow of Bilinear Transduction for molecular property prediction:
The Bilinear Transduction workflow implements a distinct process compared to traditional inductive learning. For solid-state materials, the approach typically utilizes stoichiometry-based representations to capture compositionally driven property variation [2]. For molecular systems, inputs commonly consist of molecular graphs encoded as SMILES (Simplified Molecular Input Line Entry System) representations or related formats [2] [22]. The model learns analogical input-target relations across training and test sets, enabling generalization beyond the training target support [2] [1].
Experimental Protocols and Benchmarking Standards
Comprehensive evaluation of Bilinear Transduction involves multiple benchmark datasets spanning both solid-state materials and molecular systems [2] [1]. For solids, common benchmarks include AFLOW (containing material property values from high-throughput calculations), Matbench (an automated leaderboard for benchmarking ML algorithms on solid material properties), and the Materials Project (providing materials and properties derived from high-throughput calculations) [2]. For molecular systems, datasets from MoleculeNet are frequently employed, covering graph-to-property prediction tasks including ESOL (aqueous solubility), FreeSolv (hydration free energies), Lipophilicity (octanol/water distribution coefficients), and BACE (binding affinities) [2].
Performance evaluation typically focuses on extrapolation capability measured by mean absolute error (MAE) for OOD predictions [2] [1]. Additional metrics include extrapolative precision (measuring the fraction of true top OOD candidates correctly identified) and recall of high-performing candidates [2]. Proper benchmarking requires carefully designed train-test splits that ensure test samples represent genuine OOD cases with property values outside the training distribution [2] [1].
Performance Comparison: Bilinear Transduction vs. Alternative Methods
Solid-State Materials Property Prediction
Table 1: Performance comparison (Mean Absolute Error) for solid-state materials property prediction on AFLOW dataset
| Property | Ridge Regression | MODNet | CrabNet | Bilinear Transduction |
|---|---|---|---|---|
| Band Gap [eV] | 2.59 ± 0.03 | 2.65 ± 0.04 | 1.47 ± 0.03 | 1.51 ± 0.04 |
| Bulk Modulus [GPa] | 74.0 ± 3.8 | 93.06 ± 3.7 | 59.25 ± 3.2 | 47.4 ± 3.4 |
| Debye Temperature [K] | 0.45 ± 0.03 | 0.62 ± 0.03 | 0.38 ± 0.02 | 0.31 ± 0.02 |
| Shear Modulus [GPa] | 0.69 ± 0.03 | 0.78 ± 0.04 | 0.55 ± 0.02 | 0.42 ± 0.02 |
| Thermal Conductivity [W/mK] | 1.07 ± 0.05 | 1.5 ± 0.05 | 0.97 ± 0.03 | 0.83 ± 0.04 |
Table 2: Performance comparison for materials property prediction across multiple benchmarks
| Dataset | Property | Ridge Regression | MODNet | CrabNet | Bilinear Transduction |
|---|---|---|---|---|---|
| Matbench | Band Gap [eV] | 6.37 ± 0.28 | 3.26 ± 0.13 | 2.70 ± 0.13 | 2.54 ± 0.16 |
| Matbench | Refractive Index | 14.4 ± 2.0 | 4.24 ± 0.48 | 3.92 ± 0.5 | 3.81 ± 0.49 |
| Matbench | Yield Strength [MPa] | 972 ± 34 | 731 ± 82 | 740 ± 49 | 591 ± 62 |
| Materials Project | Bulk Modulus [GPa] | 151 ± 14 | 60.1 ± 3.9 | 57.8 ± 4.2 | 45.8 ± 3.9 |
Bilinear Transduction consistently outperforms or performs comparably to established baseline methods across diverse materials property prediction tasks [2] [1]. The method demonstrates particular strength in predicting mechanical properties like bulk modulus and shear modulus, where it achieves significant reductions in MAE compared to alternatives [1]. Quantitative analysis reveals that Bilinear Transduction improves extrapolative precision by 1.8× for materials and boosts recall of high-performing candidates by up to 3× compared to conventional approaches [2].
Molecular Property Prediction
For molecular systems, Bilinear Transduction has demonstrated similar advantages in OOD prediction tasks [2]. When evaluated on molecular property prediction benchmarks, the method shows improved extrapolation capability with 1.5× better extrapolative precision for molecules compared to traditional approaches [2]. The true positive rate of OOD classification improves by 2.5× for molecules with precision improvements of 1.5× compared to non-transductive baselines [1].
The performance advantages appear most pronounced in challenging extrapolation scenarios where the target property values substantially exceed the ranges observed in training data [2]. This capability is particularly valuable for discovery-oriented research where identifying exceptional materials or molecules with unprecedented properties is the primary objective [2] [1].
Research Reagent Solutions: Essential Tools for Implementation
Table 3: Key research reagents and computational tools for implementing bilinear transduction
| Tool/Dataset | Type | Purpose | Access |
|---|---|---|---|
| MatEx | Software Library | Implementation of Bilinear Transduction for materials | https://github.com/learningmatter-mit/matex [2] |
| AFLOW | Dataset | Material properties from high-throughput calculations | Public database [2] |
| Matbench | Benchmark | Automated leaderboard for material property prediction | Public benchmark [2] |
| Materials Project | Dataset | Computed materials properties and crystal structures | Public database [2] |
| MoleculeNet | Benchmark | Molecular property prediction tasks | Public benchmark [2] |
| SMILES | Representation | Molecular structure encoding | Standard chemical notation [22] |
| COCOA | Algorithm | Compositional conservatism with anchor-seeking | https://github.com/runamu/compositional-conservatism [21] |
Successful implementation of Bilinear Transduction requires appropriate computational frameworks and datasets. The MatEx (Materials Extrapolation) library provides an open-source implementation specifically designed for OOD property prediction in materials and molecules [2]. For molecular representation, SMILES strings serve as the fundamental input format, with potential enhancements through positional embeddings in transformer architectures [22].
Recent advancements include integration with reinforcement learning frameworks through approaches like COmpositional COnservatism with Anchor-seeking (COCOA), which combines Bilinear Transduction with learned reverse dynamics models to encourage conservatism in the compositional input space [21]. This integration has demonstrated improved performance in offline reinforcement learning benchmarks, suggesting promising avenues for further development in molecular and materials design [21].
Bilinear Transduction represents a significant advancement in transductive learning approaches for zero-shot property prediction, directly addressing the critical challenge of out-of-distribution robustness in molecular property predictors [2] [1]. By reformulating the prediction problem from absolute property estimation to relative difference calculation, the method enables more accurate identification of high-performing materials and molecules with exceptional properties [2].
The consistent performance advantages demonstrated across diverse benchmark datasets suggest that Bilinear Transduction and related transductive approaches offer a promising path forward for discovery-oriented research [2] [1]. However, proper implementation requires careful attention to potential data leakage issues that can inflate performance metrics in transductive settings [7]. Future research directions likely include integration with large language models for molecular representation [22], application to emerging challenges in drug-target interaction prediction [7], and development of more sophisticated anchor selection strategies [21].
As the field progresses, transductive learning approaches like Bilinear Transduction are poised to play an increasingly important role in accelerating the discovery of novel materials and molecules with exceptional properties, potentially transforming early-stage discovery workflows across materials science and drug development [2] [1].
In the field of drug discovery, molecular property prediction models play a crucial role in prioritizing compounds for experimental validation. However, a significant limitation persists: these models typically demonstrate strong performance on compounds similar to those in their training data (in-distribution, or ID) but suffer substantial performance degradation when applied to novel, structurally distinct compounds (out-of-distribution, or OOD). This covariate shift problem is particularly problematic in real-world discovery pipelines, where the most valuable compounds for advancing research often lie beyond the chemical space represented in training datasets [23]. The fundamental challenge stems from the scarcity of labeled data, as experimental validation remains costly and time-consuming, resulting in training sets that are both small and biased toward narrow regions of chemical space.
The evaluation of OOD robustness has emerged as a critical focus in machine learning research. Heuristic assessments often lead to biased conclusions about model generalizability, as many supposedly "OOD" tests actually reflect interpolation rather than true extrapolation, potentially overestimating both generalizability and the benefits of model scaling [24]. This review compares contemporary strategies for improving OOD generalization in molecular property prediction, with particular emphasis on meta-learning approaches that leverage abundant unlabeled data to "densify" scarce labeled distributions and bridge the ID-OOD performance gap.
Methodological Comparison: Strategies for OOD Generalization
Featured Approach: Meta-Learning with Scarce Data Densification
A novel meta-learning framework addresses OOD generalization by explicitly interpolating the scarce labeled training distribution with abundant unlabeled data. This approach utilizes a permutation-invariant learnable set function, or "mixer," that combines labeled training points with context points from the unlabeled dataset. The method operates through two core components: (1) a standard meta-learner (MLP) that maps input data to feature representations, and (2) the learnable set function that mixes labeled and unlabeled representations at a specific layer. This densification strategy encourages the model to generalize more robustly under covariate shift by effectively expanding the training distribution toward regions of chemical space represented in the unlabeled data [23].
The meta-learning process employs a context set () and meta-validation set (𝒟contextcontext\mathcal{D}{\text{context}}) drawn from the unlabeled pool, enabling the model to learn an interpolation function that improves generalization to OOD compounds. This approach is particularly valuable in drug discovery settings where advancing research requires predictions about compounds with substantial distributional shifts from known molecules.𝒟mvalidmvalid\mathcal{D}{\text{mvalid}}
Alternative Paradigms for OOD Generalization
Context-Informed Heterogeneous Meta-Learning
Another advanced meta-learning approach for few-shot molecular property prediction employs a heterogeneous architecture that extracts both property-shared and property-specific molecular features. This method utilizes graph neural networks combined with self-attention encoders to capture contextual information, with an adaptive relational learning module that infers molecular relations based on shared features. The framework employs a heterogeneous meta-learning strategy where property-specific features update within individual tasks (inner loop) while all parameters update jointly (outer loop). This division enables more effective capture of both general and contextual information, leading to significant improvements in predictive accuracy, especially with limited training samples [25].
Semi-Supervised Learning with Multi-Mode Augmentation
Beyond meta-learning, enhanced semi-supervised learning (SSL) methods offer alternative pathways for leveraging unlabeled data. One approach addresses limitations of traditional SSL in small-sample environments through multi-mode augmentation, combining intra-class random augmentation with inter-class mixed augmentation. This strategy simultaneously improves both intra-class and inter-class sample completeness, creating more robust feature representations. The method incorporates an uncertainty-aware pseudo-label selection mechanism based on model prediction statistics, improving pseudo-label quality while maximizing retention of unlabeled samples. When combined with exponential moving average techniques, this approach demonstrates strong performance even with extremely limited labeled and unlabeled data [26].
Comparative Performance Analysis
Table 1: Comparative Performance of OOD Generalization Methods
| Method | Approach Category | Key Mechanism | Reported Performance Advantage | Data Requirements |
|---|---|---|---|---|
| Meta-Learning with Densification [23] | Meta-learning | Interpolates labeled data with unlabeled context points | Significant gains over SOTA under substantial covariate shift | Scarce labeled + abundant unlabeled |
| Context-Informed Heterogeneous Meta-Learning [25] | Few-shot learning | Separates property-shared and property-specific features | Superior few-shot accuracy, especially with minimal samples | Few-shot setting |
| Multi-Mode Augmentation SSL [26] | Semi-supervised learning | Combines intra-class and inter-class augmentation | Outperforms MixMatch, UDA, FreeMatch on STL-10/CIFAR-10 | Limited labeled and unlabeled data |
| Traditional Supervised Baselines | Supervised learning | Standard empirical risk minimization | Poor OOD performance due to covariate shift | Large labeled datasets |
Table 2: OOD Evaluation Metrics and Method Characteristics
| Method | Evaluation Paradigm | Handles Distribution Shifts | Main Advantages | Limitations |
|---|---|---|---|---|
| Meta-Learning with Densification [23] | OOD performance testing | Yes, via explicit interpolation | Actively densifies training distribution | Complex training pipeline |
| Heterogeneous Meta-Learning [25] | OOD performance testing | Yes, through contextual modeling | Excellent in few-shot scenarios | Requires task structure for meta-learning |
| Multi-Mode Augmentation SSL [26] | OOD performance testing | Yes, via diverse augmentation | Works with very limited data | Domain-specific augmentations needed |
| Heuristic OOD Evaluation [24] | OOD performance prediction | No, primarily for assessment | Reveals true extrapolation capability | Evaluation method, not solution |
Experimental Protocols and Methodological Details
Meta-Learning Densification Framework
The experimental protocol for the meta-learning densification approach involves several carefully designed components. The method addresses molecular property prediction under covariate shift with a small labeled dataset and abundant unlabeled molecules 𝒟train={(xi,yi)}i=1ntrain\mathcal{D}{\text{train}}={(x{i},y{i})}{i=1}^{n}caligraphicD startPOSTSUBSCRIPT train endPOSTSUBSCRIPT = { ( italicx startPOSTSUBSCRIPT italici endPOSTSUBSCRIPT , italicy startPOSTSUBSCRIPT italici endPOSTSUBSCRIPT ) } startPOSTSUBSCRIPT italici = 1 endPOSTSUBSCRIPT startPOSTSUPERSCRIPT italicn endPOSTSUPERSCRIPT. The goal is to learn a predictive model 𝒟unlabeled={xj}j=1munlabeled\mathcal{D}{\text{unlabeled}}={x{j}}{j=1}^{m}caligraphicD startPOSTSUBSCRIPT unlabeled endPOSTSUBSCRIPT = { italicx startPOSTSUBSCRIPT italicj endPOSTSUBSCRIPT } startPOSTSUBSCRIPT italicj = 1 endPOSTSUBSCRIPT startPOSTSUPERSCRIPT italicm endPOSTSUPERSCRIPT that generalizes to a distributionally shifted test set f:𝒳→𝒴f:\mathcal{X}\to\mathcal{Y}italicf : caligraphicX → caligraphicY [23].𝒟testtest\mathcal{D}{\text{test}}caligraphicD startPOSTSUBSCRIPT test endPOSTSUBSCRIPT
The core innovation lies in the mixing function, which learns to combine each labeled data point μλ\mu{\lambda}italicμ startPOSTSUBSCRIPT italicλ endPOSTSUBSCRIPT with a variable number of context points xi∼𝒟traintrainx{i}\sim\mathcal{D}{\text{train}}italicx startPOSTSUBSCRIPT italici endPOSTSUBSCRIPT ∼ caligraphicD startPOSTSUBSCRIPT train endPOSTSUBSCRIPT drawn from the unlabeled pool. For each minibatch {cij}j=1mi∼𝒟contextcontext{c{ij}}{j=1}^{m{i}}\sim\mathcal{D}{\text{context}}{ italicc startPOSTSUBSCRIPT italici italicj endPOSTSUBSCRIPT } startPOSTSUBSCRIPT italicj = 1 endPOSTSUBSCRIPT startPOSTSUPERSCRIPT italicm startPOSTSUBSCRIPT italici endPOSTSUBSCRIPT endPOSTSUPERSCRIPT ∼ caligraphicD startPOSTSUBSCRIPT context endPOSTSUBSCRIPT, the number of context points BBitalicB follows a discrete uniform distribution mim{i}italicm startPOSTSUBSCRIPT italici endPOSTSUBSCRIPT, where mi∼𝒰int(0,M)intm{i}\sim\mathcal{U}{\text{int}}(0,M)italicm startPOSTSUBSCRIPT italici endPOSTSUBSCRIPT ∼ caligraphicU startPOSTSUBSCRIPT int endPOSTSUBSCRIPT ( 0 , italicM ) controls the maximum context samples per minibatch. The mixing operation occurs at a specific layer MMitalicM, producing enriched representations lmixmixl{\text{mix}}italicl startPOSTSUBSCRIPT mix endPOSTSUBSCRIPT that incorporate information from both labeled and unlabeled distributions [23].x~i(lmix)=μλ({xi(lmix),Ci(lmix)})mixmixmix\tilde{x}{i}^{(l{\text{mix}})}=\mu{\lambda}({x{i}^{(l{\text{mix}})},C{i% }^{(l{\text{mix}})}})over~ startARG italicx endARG startPOSTSUBSCRIPT italici endPOSTSUBSCRIPT startPOSTSUPERSCRIPT ( italicl startPOSTSUBSCRIPT mix endPOSTSUBSCRIPT ) endPOSTSUPERSCRIPT = italicμ startPOSTSUBSCRIPT italicλ endPOSTSUBSCRIPT ( { italicx startPOSTSUBSCRIPT italici endPOSTSUBSCRIPT startPOSTSUPERSCRIPT ( italicl startPOSTSUBSCRIPT mix endPOSTSUBSCRIPT ) endPOSTSUPERSCRIPT , italicC startPOSTSUBSCRIPT italici endPOSTSUBSCRIPT startPOSTSUPERSCRIPT ( italicl startPOSTSUBSCRIPT mix endPOSTSUBSCRIPT ) endPOSTSUPERSCRIPT } )
Diagram 1: Meta-Learning Densification Workflow. Illustrates how labeled and unlabeled data interact through the mixing layer to produce OOD-resistant predictions.
Heterogeneous Meta-Learning Protocol
The context-informed few-shot learning approach employs a dual-component architecture where graph neural networks extract property-specific molecular features while self-attention encoders capture property-shared characteristics. The experimental protocol involves an adaptive relational learning module that infers molecular relations based on shared features. The heterogeneous meta-learning strategy implements a two-loop optimization process: inner-loop updates refine property-specific features within individual tasks, while outer-loop updates jointly optimize all parameters across tasks [25].
This approach is evaluated under rigorous few-shot learning scenarios on real molecular datasets from MoleculeNet, with performance measured through metrics such as mean absolute error and coefficient of determination (R²) for regression tasks, demonstrating superior performance compared to alternatives, particularly when training samples are severely limited [25].
Evaluation Metrics for OOD Generalization
Robust evaluation of OOD generalization requires moving beyond heuristic assessments. Current research emphasizes that many supposedly "OOD" tests actually reflect interpolation rather than true extrapolation, potentially leading to overestimated generalizability [24]. Proper OOD evaluation aims not only to assess whether a model's OOD capability is strong but also to characterize the types of distribution shifts a model can effectively address and identify safe versus risky input regions [27].
Established evaluation paradigms include OOD performance testing (with test data), OOD performance prediction (without test data), and OOD intrinsic property characterization. For molecular property prediction, metrics like mean absolute error (MAE) and coefficient of determination (R²) are commonly employed, with R² being particularly valuable as a dimensionless accuracy measure that can be compared across different OOD test sets [24].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for OOD Molecular Property Prediction
| Research Reagent | Function | Example Implementation |
|---|---|---|
| Learnable Set Function (Mixer) | Interpolates labeled and unlabeled distributions | Permutation-invariant function μ_λ [23] |
| Graph Neural Networks | Encodes molecular structure information | GIN, Pre-GNN [25] |
| Self-Attention Encoders | Captures property-shared features | Transformer-based architectures [25] |
| Multi-Mode Augmentation | Enhances sample diversity | Random + mixed augmentation strategies [26] |
| Meta-Learning Framework | Enables few-shot adaptation | MAML-inspired algorithms [23] [25] |
| Uncertainty-Aware Selection | Improves pseudo-label quality | Confidence-based filtering [26] |
The integration of meta-learning strategies with unlabeled data densification represents a promising direction for addressing the fundamental challenge of OOD generalization in molecular property prediction. By actively leveraging abundant unlabeled molecular data to expand the effective training distribution, these approaches mitigate the covariate shift problems that plague traditional supervised methods. The comparative analysis presented herein demonstrates that methods like meta-learning densification and heterogeneous meta-learning consistently outperform conventional approaches, particularly in challenging few-shot scenarios and under significant distribution shifts.
Future research directions should focus on developing more sophisticated interpolation strategies, improving the scalability of meta-learning approaches to extremely large unlabeled datasets, and establishing more rigorous OOD evaluation benchmarks that accurately distinguish between interpolation and true extrapolation. As these methodologies mature, they hold significant potential for accelerating drug discovery by providing more reliable predictions for novel compound classes that diverge from established chemical spaces.
Molecular property prediction stands as a critical task in computational chemistry and drug discovery, where accurately forecasting properties like toxicity, solubility, or bioactivity can dramatically accelerate materials research and therapeutic development. Traditional Graph Neural Networks (GNNs) have emerged as powerful tools for this task, operating directly on the graph structure of molecules where atoms represent nodes and bonds represent edges. However, these standard models face significant challenges in real-world applications where they must generalize to molecular structures and property values beyond their training distribution—a capability known as out-of-distribution (OOD) robustness.
The limitations of conventional GNNs have spurred interest in more advanced architectures that better capture the physical and geometric principles governing molecular systems. Among these, E(3)-equivariant architectures and hybrid models have shown particular promise. E(3)-equivariant Graph Neural Networks explicitly embed the symmetries of 3D Euclidean space—translation, rotation, and reflection—directly into their architecture, ensuring predictions transform consistently with molecular orientation. Hybrid models combine complementary architectural paradigms, such as integrating transformer components with GNNs or incorporating quantum-inspired elements, to overcome limitations of single-architecture approaches.
This guide provides a systematic comparison of these advanced architectures, focusing on their performance, robustness, and applicability across diverse molecular prediction tasks, with particular emphasis on their OOD generalization capabilities—a crucial consideration for real-world deployment where novel molecular scaffolds are frequently encountered.
Theoretical Foundations: From Invariance to Equivariance
The Geometric Principles of E(3)-Equivariance
E(3)-equivariant networks fundamentally differ from standard GNNs through their explicit handling of 3D geometric symmetries. The E(3) group encompasses all translations, rotations, and reflections in 3D Euclidean space. For molecular systems, where properties should not depend on arbitrary orientation or placement in space, leveraging these symmetries is crucial for physical meaningfulness and data efficiency.
Equivariance refers to the property that when the input to a network undergoes a transformation (e.g., rotation), the representation at each layer transforms in a corresponding way. Formally, a function 𝑓: 𝑋 → 𝑌 is equivariant to a group 𝐺 if for any transformation 𝑔 ∈ 𝐺, 𝑓(𝑔·𝑥) = 𝑔·𝑓(𝑥). This contrasts with invariance, where 𝑓(𝑔·𝑥) = 𝑓(𝑥). For molecular systems, invariance is desired for scalar outputs like energy, while equivariance is essential for vector or tensor outputs like forces or dipole moments [28].
Standard GNNs typically achieve invariance through data augmentation or specific architectural choices, but this approach can be computationally inefficient and may fail to capture important geometric dependencies. E(3)-equivariant models like EGNNs (E(n) Equivariant Graph Neural Networks) build equivariance directly into their operations through carefully designed coordinate updates and message-passing schemes that preserve transformation properties across layers [29] [30].
Hybrid Architecture Paradigms
Hybrid architectures seek to combine the strengths of multiple approaches to overcome limitations of individual paradigms:
- Graph Transformer Hybrids: Integrate the global receptive field of transformers with the structural inductive biases of GNNs, using attention mechanisms to capture long-range dependencies in molecular graphs [31] [32].
- Quantum-Classical Hybrids: Incorporate quantum-inspired components or quantum neural networks to enhance modeling of complex quantum chemical relationships, particularly valuable for data-sparse scenarios [33].
- Multi-Scale and Multi-Fidelity Models: Combine information from different levels of theory (e.g., DFT calculations with experimental data) or different molecular representations to improve generalization [31] [34].
These hybrid approaches aim to balance the expressive power of large, general models with the sample efficiency of specialized architectures incorporating domain knowledge.
Architectural Comparison: Capabilities and Trade-offs
Table 1: Key Characteristics of Advanced Molecular Property Prediction Architectures
| Architecture Type | Key Examples | Symmetry Handling | Molecular Representation | Key Advantages |
|---|---|---|---|---|
| E(3)-Equivariant GNNs | EGNN [30], EquiPPIS [29] | E(3)-equivariant | 3D coordinates + graph | Native geometric awareness; data efficient; robust to rotations |
| Graph Transformer Hybrids | Graphormer [30], CrysCo [31] | Permutation equivariant + encodings | Graph + positional encodings | Global attention; strong on large molecules; excellent benchmarks |
| Quantum-Hybrid Models | PolyQT [33] | Varies with base architecture | SMILES/Graph + quantum components | Strong on sparse data; captures complex nonlinearities |
| Meta-Learning Architectures | CFS-HML [34] | Property-specific encoders | Multi-task graph representations | Excellent few-shot performance; adapts to new properties |
Performance Analysis Across Property Types
Table 2: Quantitative Performance Comparison Across Molecular Property Types
| Architecture | Quantum Properties (QM9 MAE) | Environmental Fate (LogKow MAE) | Bioactivity (MolHIV ROC-AUC) | OOD Generalization (Avg. Error vs. ID) |
|---|---|---|---|---|
| EGNN | 0.15-0.35 (varies by target) [30] | 0.22 (logK_d) [30] | 0.781 [30] | 3.0× ID error [9] |
| Graphormer | 0.18-0.40 (varies by target) [30] | 0.18 (logKow) [30] | 0.807 [30] | Not reported |
| EquiPPIS (Specialized) | N/A (PPI prediction) | N/A | N/A | Better with AF2 models than competing methods with experimental structures [29] |
| CFS-HML (Few-shot) | Not reported | Not reported | ~6% improvement over baselines in few-shot [34] | Not systematically evaluated |
The performance data reveals several important patterns. First, problem-domain fit significantly influences architectural effectiveness. EGNN demonstrates strong performance on geometry-sensitive properties like environmental partition coefficients (logK_d MAE: 0.22), leveraging its inherent 3D coordinate processing [30]. Graphormer excels on tasks requiring global reasoning across molecular structures, achieving the highest reported accuracy on logKow prediction (MAE: 0.18) and bioactivity classification (ROC-AUC: 0.807) on the OGB-MolHIV dataset [30].
Critically, the BOOM benchmark for OOD molecular property prediction reveals that even the top-performing models exhibit an average OOD error 3× larger than in-distribution error [9] [11]. This performance gap highlights the substantial challenge of OOD generalization in molecular machine learning and underscores why robustness should be a primary consideration in architecture selection.
Specialized architectures like EquiPPIS demonstrate that properly encoding physical symmetries can yield remarkable robustness—the model attains better accuracy with AlphaFold2-predicted structural models than what existing methods achieve with experimental structures [29].
Experimental Protocols and Methodologies
Benchmarking Out-of-Distribution Generalization
The BOOM benchmark establishes rigorous methodology for evaluating OOD performance in molecular property prediction [9] [11]. Rather than partitioning data randomly, BOOM creates OOD splits based on property value distributions, selecting molecules with the lowest probability densities (tail ends of distribution) for the OOD test set. This approach directly aligns with the molecule discovery goal of identifying compounds with novel property values.
Key aspects of the BOOM protocol include:
- Using kernel density estimators to identify low-probability regions of property space
- Allocating lowest 10% of probability scores to OOD set (for QM9 dataset)
- Maintaining identical model architectures and training procedures between ID and OOD evaluations
- Evaluating across multiple molecular representations (SMILES, graphs) and model types
This methodology reveals that while models with high inductive bias (like geometrically-informed GNNs) can perform well on OOD tasks with simple, specific properties, current chemical foundation models surprisingly do not show strong OOD extrapolation capabilities [9].
Equivariant Architecture Implementation
The core innovation in E(3)-equivariant models like EGNN lies in their message passing and coordinate update schemes [30]. The typical implementation involves:
- Graph Construction: Molecules represented as graphs with node features 𝒉ᵢ and coordinates 𝒙ᵢ
- Equivariant Message Passing: Messages between nodes computed using relative displacements and distances
- Coordinate Updates: Node coordinates updated using rotationally-equivariant functions of incoming messages
- Invariant Node Updates: Node features updated using invariant aggregation of messages
This design ensures that rotations or translations of input coordinates result in corresponding transformations of internal representations and outputs, without requiring data augmentation or losing geometric information through invariant featurization.
Hybrid Model Training Approaches
Hybrid architectures often employ sophisticated training schemes to balance different components:
CrysCo Framework (for materials property prediction) utilizes parallel networks—a crystal GNN (CrysGNN) and composition-based transformer (CoTAN)—trained jointly in a hybrid manner [31]. The model incorporates four-body interactions (atoms, bonds, angles, dihedrals) through multiple graph representations, explicitly capturing periodicity and structural characteristics of crystalline materials.
CFS-HML employs heterogeneous meta-learning with separate optimization loops for property-shared and property-specific knowledge encoders [34]. The inner loop updates property-specific parameters on individual tasks, while the outer loop jointly updates all parameters across tasks, enabling effective few-shot learning.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools and Datasets for Molecular Property Prediction Research
| Tool/Dataset | Type | Primary Function | Relevance to Advanced Architectures |
|---|---|---|---|
| QM9 [9] [30] | Dataset | 134k small organic molecules with quantum chemical properties | Benchmarking quantum property prediction; standard for 3D molecular tasks |
| OGB-MolHIV [30] | Dataset | ~41k molecules for HIV replication inhibition prediction | Evaluating bioactivity prediction on realistic drug discovery task |
| BOOM Benchmark [9] [11] | Evaluation Framework | Standardized OOD testing protocols | Critical for assessing real-world robustness of new architectures |
| DeePMD-kit [28] | Software | Deep potential molecular dynamics implementation | Production-scale equivariant model training for molecular dynamics |
| ALIGNN [31] | Architecture | GNN with angle information in message passing | Incorporates higher-order geometric interactions (3-body, 4-body) |
Critical Analysis and Future Directions
Architectural Trade-offs and Selection Guidelines
The comparative analysis reveals several key trade-offs that should guide architecture selection:
E(3)-equivariant models (EGNN, EquiPPIS) excel when 3D structural information is available and geometrically sensitive predictions are needed, offering strong OOD generalization from limited data due to their physical inductive biases [29] [30]. They are particularly valuable for protein-protein interaction prediction, quantum property estimation, and conformation-dependent tasks.
Graph Transformer hybrids (Graphormer, CrysCo) demonstrate superior performance on tasks requiring global reasoning across molecular structures and when leveraging large-scale datasets [31] [30]. Their attention mechanisms effectively capture long-range dependencies in molecular graphs.
Meta-learning approaches (CFS-HML) show exceptional promise for low-data scenarios and multi-property prediction, adaptively balancing property-shared and property-specific knowledge [34]. These are ideal for early-stage discovery where labeled data is scarce for specific properties.
Quantum-hybrid models (PolyQT) offer intriguing capabilities for modeling complex nonlinear relationships, particularly evident in polymer informatics where they maintain performance even under significant data sparsity [33].
Frontier Challenges and Emerging Solutions
Despite considerable advances, significant challenges remain at the frontier of molecular property prediction:
OOD Generalization continues to present the most significant hurdle, with even state-of-the-art models showing substantially increased error (3×) on OOD samples [9]. Promising directions include:
- Developing more sophisticated distribution-shift benchmarks covering diverse molecular families
- Integrating active learning and model-data co-design frameworks to strategically expand training distributions
- Creating foundation models with explicit OOD generalization objectives rather than merely optimizing in-distribution performance
Data Fidelity and Multi-Fidelity Learning represents another critical challenge. Current models are ultimately limited by the quality and diversity of their training data [28]. Transfer learning from data-rich source tasks (e.g., formation energy prediction) to data-scarce target tasks (e.g., mechanical property prediction) shows promise for addressing data scarcity [31].
Interpretability and Explainability remain crucial for scientific adoption, particularly as models grow more complex. Emerging techniques that provide insight into model decision-making, such as attention visualization in transformer hybrids or contribution analysis in equivariant networks, will be essential for building trust and extracting scientific insight [31] [28].
The integration of physical principles through specialized architectures like E(3)-equivariant networks, combined with the representational power of hybrid models, points toward a future where molecular property predictors achieve both high accuracy and robust generalization—ultimately accelerating the discovery of novel materials and therapeutics.
The application of deep learning to molecular discovery promises to accelerate the identification of novel materials and therapeutics. However, the ultimate utility of these models depends on their ability to make accurate predictions for out-of-distribution (OOD) molecules—those with property values or structural scaffolds not represented in the training data [9]. Discovery inherently requires venturing beyond known chemical space, making OOD generalization a frontier challenge in chemical machine learning [9]. Among the various model architectures being explored, transformer-based models, pre-trained on large chemical databases, are emerging as a powerful class of chemical foundation models. This guide provides an objective comparison of the OOD performance of key transformer models, including MolFormer and ChemBERTa, situating them within the broader landscape of molecular property predictors.
Performance Comparison of Molecular Property Predictors
The following tables synthesize quantitative performance data from large-scale benchmark studies, primarily the BOOM (Benchmarking Out-Of-distribution Molecular property predictions) benchmark, which evaluated over 140 model and task combinations [9].
Table 1: Overview of Model Architectures and OOD Performance
| Model Name | Architecture Type | Molecular Representation | Key OOD Finding | Avg. OOD Error vs. ID |
|---|---|---|---|---|
| MolFormer | Transformer (T5 backbone) | SMILES | Does not show strong OOD extrapolation [9]. | N/A |
| ChemBERTa | Transformer (BERT backbone) | SMILES | Does not show strong OOD extrapolation [9]. | N/A |
| ModernBERT | Transformer (Modern architecture) | SMILES | Does not show strong OOD extrapolation [9]. | N/A |
| Random Forest | Traditional ML | RDKit Descriptors | Baseline model; outperformed by some GNNs on specific OOD tasks [9]. | N/A |
| Chemprop | Graph Neural Network (GNN) | Molecular Graph | Can perform well on OOD tasks with simple, specific properties [9]. | Varies by task |
| IGNN | GNN (Invariant) | Molecular Graph + Distances | High inductive bias can aid in specific OOD tasks [9]. | Varies by task |
| Bilinear Transduction | Transductive Model | Stoichiometry/Graph | Improves extrapolation precision for materials (1.8×) and molecules (1.5×) [2]. | Lower MAE than baselines [2] |
Table 2: Detailed OOD Performance on QM9 Molecular Property Datasets Data from the BOOM benchmark, which defined OOD based on tail-end property values [9].
| Property (Dataset) | Top Performing Model(s) | OOD Performance Notes |
|---|---|---|
| Isotropic Polarizability (α) | Not Specified | Even top-performing models showed an average OOD error 3x larger than in-distribution (ID) error [9]. |
| HOMO-LUMO Gap | Not Specified | No single model achieved strong OOD generalization across all 10 benchmarked tasks [9]. |
| Dipole Moment (μ) | Not Specified | Deep learning models with high inductive bias (e.g., certain GNNs) performed well on OOD tasks with simple properties [9]. |
| Heat Capacity (Cv) | Not Specified | Current chemical foundation models (including transformers) did not demonstrate strong OOD extrapolation capabilities [9]. |
Experimental Protocols for OOD Benchmarking
The BOOM Benchmark Methodology
A key methodology for evaluating OOD generalization in the chemical domain is the BOOM benchmark [9]. Its experimental protocol is detailed below.
Workflow: BOOM OOD Benchmarking
- OOD Splitting Strategy: The BOOM benchmark defines OOD with respect to the model's output—the molecular property values. For a given property dataset, a Kernel Density Estimator (KDE) with a Gaussian kernel is fitted to the distribution of property values. Each molecule is assigned a probability score based on this KDE. The OOD test set is constructed from the molecules with the lowest probability scores (e.g., the lowest 10% for the QM9 dataset), which correspond to the tail ends of the property value distribution. This method directly aligns with the goal of discovering molecules with extreme, novel properties [9].
- Datasets: The benchmark utilizes 10 molecular property datasets. Eight are from the QM9 dataset, which includes 133,886 small organic molecules (CHONF) and properties like HOMO-LUMO gap and dipole moment calculated via Density Functional Theory (DFT). The other two (density and solid heat of formation) are from the 10k Dataset, derived from experimentally synthesized CHON molecules in the Cambridge Crystal Structure Dataset [9].
- Evaluation: Models are evaluated based on their prediction error (e.g., Mean Absolute Error) on the held-out OOD test set and compared against their performance on a randomly sampled in-distribution (ID) test set.
Real-World MOOD Generalization Protocol
Another critical protocol focuses on Molecular Out-Of-Distribution (MOOD) generalization, which characterizes the covariate shifts encountered in real-world drug discovery [35].
- Splitting Strategy: This approach defines OOD based on the input (molecular structure). It involves splitting data such that the training and test sets are separated by a significant distance in the chemical representation space. Common methods include scaffold splitting (separating molecules based on their Bemis-Murcko scaffold) and more challenging cluster-based splits (using chemical similarity clustering like K-means on molecular fingerprints) [35] [16].
- Performance Metrics: Beyond prediction error, this protocol emphasizes the drop in performance and uncertainty calibration between ID and OOD sets. Real-world shifts have been shown to cause performance drops of up to 60% and miscalibration by up to 40% [35].
The Scientist's Toolkit: Research Reagents & Computational Solutions
Table 3: Essential Computational Tools for OOD Molecular Property Prediction
| Tool / Resource | Type | Primary Function in OOD Research |
|---|---|---|
| BOOM Benchmark | Software Benchmark | Provides a standardized methodology and dataset splits for evaluating OOD generalization of property prediction models [9]. |
| QM9 Dataset | Molecular Dataset | A standard dataset of small organic molecules and their quantum mechanical properties for training and benchmarking models [9]. |
| RDKit | Open-Source Toolkit | Used to generate molecular descriptors and fingerprints, which serve as features for traditional machine learning models and for analyzing chemical space [9] [36]. |
| ChemBERTa / MolFormer | Pre-trained Models | Transformer-based foundation models that can be fine-tuned on specific property prediction tasks to assess their OOD transfer capabilities [9]. |
| Conformal Prediction | Statistical Framework | A method (e.g., TESSERA) to provide per-sample prediction intervals with coverage guarantees, improving reliability under distribution shift [37]. |
Discussion and Synthesis of Findings
The experimental data leads to several key conclusions regarding the OOD capabilities of transformer-based chemical foundation models:
- Overall OOD Performance Gap: Large-scale benchmarks reveal that no current model, including transformer-based foundation models, achieves strong OOD generalization across a wide range of tasks [9]. In the BOOM benchmark, even the top-performing model exhibited an average OOD error that was three times larger than its ID error. This underscores OOD generalization as a significant, unsolved challenge in the field.
- Transformers vs. Other Architectures: While transformers like ChemBERTa and MolFormer have shown impressive in-distribution performance, the current evidence suggests they do not yet demonstrate superior OOD extrapolation capabilities compared to other architectures. Their performance is context-dependent, and they have not consistently outperformed models with strong inductive biases, such as Graph Neural Networks (GNNs) designed for molecular graphs, on OOD tasks [9].
- The Impact of Splitting Strategy: The perceived performance of a model is highly sensitive to how OOD data is defined. Studies show that while models may maintain reasonable performance on scaffold splits, they face a much harder challenge on cluster-based splits where the chemical similarity between training and test sets is more rigorously controlled [16]. This highlights the necessity of choosing an OOD benchmark that reflects the intended real-world application.
- Promising Research Directions: Several approaches show promise for improving OOD robustness. Transductive methods like Bilinear Transduction, which learns from analogical input-target relations, have demonstrated improved extrapolation precision [2]. Uncertainty quantification methods like TESSERA, which leverage Mixture of Experts and conformal prediction, provide more reliable and adaptive prediction intervals under distribution shift [37]. Furthermore, novel training paradigms that leverage unlabeled data to densify the space between ID and OOD regions are also being explored [38].
In summary, the assessment of chemical foundation models like MolFormer and ChemBERTa reveals a critical performance gap when faced with out-of-distribution data. Despite their power and pre-training on vast chemical datasets, these transformers have not yet proven to be a definitive solution for OOD generalization in molecular property prediction. The choice of model architecture should be guided by the specific property task and the nature of the expected distribution shift. For researchers and drug development professionals, this emphasizes the importance of rigorous OOD benchmarking using protocols like BOOM and MOOD before deploying models in discovery pipelines. Future progress will likely depend on architectural innovations, improved pre-training strategies, and the broader adoption of specialized OOD techniques like transduction and robust uncertainty quantification.
Diagnosing and Overcoming Common OOD Failures and Overconfident Predictions
The discovery of novel, high-performing materials and drug candidates fundamentally depends on identifying molecules with property values that fall outside known distributions, a challenge that requires machine learning (ML) models to extrapolate rather than merely interpolate [2]. This challenge is exacerbated by covariate shift, a phenomenon where the distribution of input variables (molecular features) differs between the training and test datasets [39] [40]. In drug discovery, covariate shift frequently occurs when a model trained on one chemical series must predict on a new, structurally distinct series, compromising prediction accuracy and hindering the identification of promising candidates [39]. The core problem is that standard ML models presume training and test data are independently and identically distributed (i.i.d.), an assumption often violated in real-world applications due to evolving chemical space exploration [39] [40].
The ability to generalize to out-of-distribution (OOD) data is a new frontier challenge in chemical machine learning [9]. When OOD generalization is defined with respect to the range of the predictive function—predicting property values beyond those seen in training—classical ML models face significant difficulties [2]. This article objectively compares emerging techniques designed to stabilize predictions across novel chemical scaffolds, framing the discussion within the broader thesis of evaluating OOD robustness in molecular property predictors.
Quantitative Comparison of OOD Performance Techniques
Systematic benchmarking studies reveal that no single model currently achieves strong OOD generalization across all molecular property prediction tasks. The BOOM (Benchmarking Out-Of-distribution Molecular property predictions) initiative evaluated over 140 model-task combinations, finding that even top-performing models exhibited an average OOD error 3x larger than their in-distribution error [9]. This section provides a structured comparison of established and emerging methodologies.
Table 1: Performance Comparison of OOD Techniques on Solid-State Materials
| Technique | Average OOD MAE Reduction | Extrapolative Precision Boost | Recall of High-Performers |
|---|---|---|---|
| Bilinear Transduction (MatEx) | Consistently lower vs. baselines [2] | 1.8× for materials [2] | Up to 3× boost [2] |
| Ridge Regression | Baseline [2] | Baseline [2] | Baseline [2] |
| MODNet | Comparable or outperformed by Bilinear Transduction [2] | Not specified | Lower than Bilinear Transduction [2] |
| CrabNet | Comparable or outperformed by Bilinear Transduction [2] | Not specified | Lower than Bilinear Transduction [2] |
Table 2: Performance of Model Architectures on Molecular OOD Tasks (Based on BOOM Benchmark)
| Model Architecture | Representative Model | Key Finding on OOD Generalization |
|---|---|---|
| Traditional ML | Random Forest (RDKit Featurizer) | Baseline performance; struggles with complex OOD tasks [9] |
| Graph Neural Network (GNN) | Chemprop, TGNN | High inductive bias can help on OOD tasks with simple, specific properties [9] |
| Transformer (Encoder-Only) | ChemBERTa | Current foundation models do not show strong OOD extrapolation capabilities [9] |
| Transformer (Encoder-Decoder) | MolFormer | Current foundation models do not show strong OOD extrapolation capabilities [9] |
| Transformer (Autoregressive) | Regression Transformer (XLNet-based) | Current foundation models do not show strong OOD extrapolation capabilities [9] |
| Equivariant GNN | EGNN, MACE | Performance varies; no model dominates across all tasks [9] |
The correlation between in-distribution (ID) and OOD performance is not guaranteed and depends heavily on the data splitting strategy. While a strong positive correlation (Pearson r ~ 0.9) exists for scaffold splitting, this correlation significantly weakens (Pearson r ~ 0.4) for the more challenging cluster-based splitting [41]. This indicates that model selection based solely on ID performance is insufficient for applications requiring OOD robustness.
Experimental Protocols and Methodologies for OOD Evaluation
Establishing Robust OOD Benchmarks
A critical prerequisite for comparing techniques is a robust methodology for evaluating OOD performance. The BOOM benchmark adopts a property-based OOD splitting strategy. For a given molecular property dataset, a kernel density estimator (with Gaussian kernel) is fitted to the property values. The OOD test set is constructed from the molecules with the lowest 10% of probability scores, effectively selecting samples at the tails of the property value distribution. The remaining molecules are used for training and in-distribution (ID) testing [9]. This approach directly aligns with the goal of discovering molecules with state-of-the-art properties that extrapolate beyond the training data.
The Bilinear Transduction Protocol (MatEx)
The Bilinear Transduction method, implemented in the MatEx (Materials Extrapolation) library, reparameterizes the prediction problem. Instead of predicting property values directly from a new material, it learns how property values change as a function of material differences [2].
Workflow:
- Reparameterization: For a target material, instead of a direct prediction, the model uses a known training example and the difference in representation space between the training and target material.
- Training: The model learns a bilinear map that captures how property differences relate to representation differences.
- Inference: Property values for a new sample are predicted based on a chosen training example and the representation space difference between that example and the new sample [2].
This method has been evaluated on benchmarks like AFLOW, Matbench, and the Materials Project, covering 12 distinct prediction tasks for electronic, mechanical, and thermal properties [2].
The Scaffold-Aware Augmentation Protocol (ScaffAug)
To address class and structural imbalance, the ScaffAug framework employs a generative augmentation approach [42].
Workflow:
- Scaffold-Aware Sampling (SAS): Identifies scaffolds from known active molecules and uses a sampling strategy to prioritize those that are underrepresented, building a balanced scaffold library.
- Scaffold Extension: Employs a graph diffusion model (DiGress) to generate novel, valid molecules that preserve the core scaffold structures from the library. This conditions the generation on chemically meaningful regions.
- Self-Training with Pseudo-Labeling: Safely integrates the generated synthetic data with the original labeled data using a confidence-based pseudo-labeling strategy.
- Diversity Reranking: Applies Maximal Marginal Relevance (MMR) to the model's top predictions to enhance scaffold diversity in the final recommended set, balancing predicted activity with structural novelty [42].
Diagram 1: The ScaffAug Framework for OOD Generalization.
Covariate Shift Identification and Correction
A foundational step in tackling covariate shift is its identification. A common technique is to treat it as a classification problem [40].
Protocol for Identifying Drifting Features:
- Preprocessing: Impute missing values and label encode categorical variables from both training and test sets.
- Create Mixed Dataset: Take random samples of equal size from the training and test data. Add a new feature,
origin, labeled astrainortest. - Model Training: Train a binary classifier (e.g., Random Forest) to predict the
originusing one feature at a time on a subset (e.g., 75%) of the mixed dataset. - Evaluate Drift: Predict on the held-out portion (25%) and calculate the AUC-ROC for each feature. A feature with an AUC-ROC greater than 0.80 is typically classified as a drifting feature, indicating its distribution differs significantly between train and test sets [40].
For correction, the Kullback-Leibler Importance Estimation Procedure (KLIEP) is a noted method that reweights instances in the training data to align its distribution more closely with the prediction set, though its practical effectiveness can vary [39].
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Key Computational Tools for OOD Molecular Property Prediction
| Tool / Solution | Function / Application | Relevance to OOD Robustness |
|---|---|---|
| MatEx (Materials Extrapolation) | Open-source implementation of Bilinear Transduction [2] | Enables zero-shot extrapolation to higher property value ranges [2] |
| BOOM Benchmark | Standardized benchmark for OOD molecular property prediction [9] | Provides robust evaluation framework across 10+ tasks and 12+ models [9] |
| ScaffAug Framework | Scaffold-aware generative augmentation and reranking [42] | Mitigates structural and class imbalance in virtual screening [42] |
| Graph Diffusion Models (e.g., DiGress) | Generation of valid molecules conditioned on scaffolds [42] | Creates structurally diverse training data for better OOD learning [42] |
| KLIEP Algorithm | Covariate shift correction via instance reweighting [39] | Adjusts training distribution to be more similar to prediction set [39] |
| RDKit Featurizer | Generates chemically-informed molecular descriptors [9] | Provides baseline features for traditional ML models in benchmarks [9] |
The systematic comparison of techniques reveals a dynamic field actively addressing the critical challenge of covariate shift. Bilinear Transduction shows marked improvement in extrapolative precision for solid-state materials, while scaffold-aware generative approaches like ScaffAug offer a promising path to overcoming data imbalance in molecular screening. A key consensus from benchmarking efforts is that no single model architecture currently dominates all OOD tasks, and performance is highly sensitive to the specific splitting strategy used for evaluation [9] [41]. The development of ML models with strong, consistent OOD generalization remains a frontier challenge, necessitating continued investment in standardized benchmarks, novel architectures with stronger inductive biases, and data generation strategies that explicitly account for scaffold diversity and property value extremes. The future of accelerated molecule and material discovery hinges on this pursuit of robustness beyond the training distribution.
The application of deep learning in molecular property prediction has revolutionized aspects of drug discovery and development. However, traditional models utilizing Softmax output functions frequently produce overconfident predictions for out-of-distribution (OOD) samples—molecules structurally dissimilar to those in the training data. This overconfidence poses a significant risk in experimental pipelines, potentially leading to misallocated resources and failed validation studies. In molecular property prediction, where chemical space exploration inherently involves venturing beyond training distributions, robust uncertainty quantification (UQ) becomes paramount for reliable decision-making.
This guide objectively compares two advanced UQ approaches—Evidential Deep Learning (EDL) and Normalizing Flows—against traditional methods. We frame this comparison within the critical research thesis of evaluating out-of-distribution robustness in molecular property predictors, providing experimental data and implementation protocols to guide researchers and drug development professionals.
Technical Comparison of UQ Methods
The table below summarizes the core characteristics, mechanisms, and comparative performance of different UQ methods relevant to molecular sciences.
Table 1: Comparison of Uncertainty Quantification Methods in Molecular Property Prediction
| Method | Core Mechanism | Uncertainty Types Captured | Computational Cost | Key Advantages | Key Limitations in OOD Scenarios |
|---|---|---|---|---|---|
| Softmax (Baseline) | Point-estimate class probabilities | Aleatoric (implicitly, often poorly) | Low | Simple, widely implemented | High overconfident errors on OOD data; poor calibration [43] |
| Bayesian Neural Networks (BNNs) | Learns parameter distributions via sampling | Epistemic & Aleatoric | Very High | Principled uncertainty decomposition | Computationally prohibitive for large screens [44] |
| Monte Carlo (MC) Dropout | Approximates BayesNet with dropout at inference | Primarily Epistemic | Medium | Easy implementation on existing models | Multiple forward passes increase inference time [45] |
| Deep Ensembles | Variance from multiple independent models | Epistemic & Aleatoric | High | Strong empirical performance; simple | High training cost; parameter storage [44] |
| Evidential Deep Learning (EDL) | Predicts parameters of a prior Dirichlet distribution | Epistemic & Aleatoric | Low | Single forward pass; fast inference | Restrictive Dirichlet assumption can limit robustness [46] |
| Normalizing Flows (in EDL) | Learns complex posterior densities in latent space | Epistemic & Aleatoric | Medium | More flexible density estimation; enhanced OOD detection | Higher complexity than standard EDL [47] [43] |
Experimental Performance and OOD Robustness
Quantitative evaluations on molecular datasets reveal significant performance differences between UQ methods, especially under challenging out-of-distribution splits.
Table 2: Empirical Performance Comparison on Molecular Property Prediction Tasks
| Study Context | Evaluation Metric | Softmax/ Baseline | Standard EDL | EDL + Normalizing Flows | Notes on OOD Setting |
|---|---|---|---|---|---|
| ADMET & LBVS Tasks [43] | Overconfident Failure Reduction | Baseline | Notable Improvement | Greatest Improvement | AttFpPost model reduced OF predictions on OOD molecules [43] |
| HiggsML Challenge [47] | Parameter Estimation Robustness | Not Applicable | Good Performance | Top Performance (1st Place) | Handles systematic uncertainties and data shifts [47] |
| DMNIST Benchmark [46] | Separation of Noisy ID/OOD Uncertainty | Substantial overlap | Limited separation | Clean separation achieved by ℱ-EDL | Illustrates enhanced expressiveness over Dirichlet-based EDL [46] |
| Lower-N QSAR Regression [44] | RMSE on Top 5% Most Certain | Higher RMSE | Competitive RMSE | N/A | EDL showed lowest error on Delaney, Freesolv, QM7 [44] |
| Cluster-Based Splitting [16] | ID-OOD Performance Correlation | Weak correlation (r~0.4) | N/A | N/A | Highlights need for rigorous OOD evaluation beyond scaffold splits [16] |
Key Experimental Protocols and Methodologies
Implementing and evaluating these UQ methods requires specific experimental designs. Below are detailed protocols for critical experiments cited in this guide.
Protocol: Evaluating OOD Robustness on Molecular Data
This protocol is based on experiments evaluating model performance on out-of-distribution data, a core challenge in molecular property prediction [43] [16].
- Objective: To assess the propensity of models to make overconfident errors on molecules outside the training distribution.
- Dataset Preparation: Use a molecular dataset (e.g., ADMET properties). Instead of random splitting, employ scaffold splitting (grouping by Bemis-Murcko scaffolds) or, for a greater challenge, chemical similarity clustering (e.g., K-means on ECFP4 fingerprints) to create train/test splits. Cluster splitting poses the hardest OOD challenge [16].
- Model Training: Train the candidate models (e.g., Softmax-based GNN, EDL model, Flow-based EDL) on the training set.
- Evaluation:
- Identify misclassified test samples.
- For these misclassifications, plot the distribution of predicted confidence (e.g., max Softmax probability, uncertainty metrics).
- A robust UQ method will show that most mispredictions have low confidence/high uncertainty. A model with poor UQ will have many mispredictions with high confidence, indicating overconfident errors [43].
- Key Output: The rate of "Overconfident False" (OF) predictions, which should be significantly lower for Flow-enhanced EDL models compared to vanilla Softmax models [43].
Protocol: Uncertainty-Guided Virtual Screening
This protocol tests the practical utility of UQ in a ligand-based virtual screening (LBVS) campaign, as demonstrated in literature [43] [44].
- Objective: To prioritize molecules for screening with higher success rates by leveraging uncertainty estimates.
- Workflow:
- Train a property predictor (e.g., for P-gp inhibition) with a robust UQ method like AttFpPost (Flow-based EDL) [43].
- Screen a large, diverse chemical library. For each molecule, obtain both the predicted property (e.g., probability of activity) and its uncertainty estimate.
- Apply a confidence threshold: Filter the ranked list to retain only molecules where the predictive uncertainty is below a predefined level.
- Evaluation: Compare the early enrichment (e.g., hit rate in the top 1% or 5% of the screened list) of the uncertainty-guided screen versus a screen based solely on predicted probability. Models with better UQ should demonstrate improved enrichment by filtering out high-uncertainty, likely erroneous predictions [43].
- Key Output: Enhanced screening power and validation rates in retrospective studies [44].
Protocol: Assessing Uncertainty Calibration
This protocol evaluates how well a model's predicted confidence aligns with its actual accuracy [43].
- Objective: To determine if a predicted probability of 0.9 corresponds to a 90% chance of being correct.
- Method:
- After model training, run inference on a held-out test set.
- Bin the test samples based on their predicted confidence (e.g., 0.0-0.1, 0.1-0.2, ..., 0.9-1.0).
- For each bin, calculate the average predicted confidence and the actual accuracy (fraction of correct predictions).
- Plot the reliability curve: accuracy vs. confidence.
- Interpretation: A perfectly calibrated model will have a diagonal reliability curve. Traditional Softmax models often show a diverging curve, indicating overconfidence. Well-calibrated EDL and Flow-based models will have curves closer to the diagonal [43].
Visualizing Workflows and Logical Relationships
EDL and Normalizing Flow Integration for Molecular Property Prediction
The following diagram illustrates the integrated workflow of an Evidential Deep Learning model enhanced with Normalizing Flows for superior uncertainty quantification.
Decision Framework for UQ Method Selection
This flowchart provides a logical pathway for researchers to select an appropriate uncertainty quantification method based on their specific project constraints and goals.
The Scientist's Toolkit: Research Reagents and Computational Solutions
This table details key software, architectural components, and data resources essential for implementing and experimenting with advanced UQ methods in molecular informatics.
Table 3: Essential Research Toolkit for UQ in Molecular Property Prediction
| Tool/Component | Type | Function in UQ Research | Example Implementations / Notes |
|---|---|---|---|
| Graph Neural Networks (GNNs) | Model Architecture | Learns molecular representations from graph structure; backbone for property prediction. | Message Passing Neural Networks (MPNNs), Attentive FP [43] |
| Dirichlet Distribution | Statistical Model | Serves as the prior in EDL; models distribution over class probabilities. | Standard in EDL; generalized by Flexible Dirichlet (FD) in ℱ-EDL [46] |
| Flexible Dirichlet (FD) | Statistical Model | Generalization of Dirichlet; allows multimodal beliefs on simplex for more expressive UQ. | Core component of ℱ-EDL; provides enhanced robustness [46] |
| Normalizing Flows | Model Component | Learns complex, invertible transformations to model intricate posterior distributions in latent space. | Used in PostNet and Contrastive NFs for density estimation [47] [43] |
| Chemical Splitting Scripts | Data Curation | Generates meaningful train/test splits to evaluate OOD robustness. | Scaffold split, cluster-based split (hardest OOD challenge) [16] |
| Evidential Loss Function | Training Algorithm | Regularizes learning to prevent overfitting and encourage evidence accumulation on seen data. | Combines prediction error (e.g., MSE) with KL divergence penalty [46] [44] |
| Uncertainty Metrics | Evaluation | Quantifies different aspects of model confidence for comparison. | Epistemic uncertainty (e.g., Var[μ]), aleatoric uncertainty (e.g., E[σ²]), predictive entropy [44] |
The transition beyond Softmax to more sophisticated uncertainty quantification methods is crucial for developing reliable molecular property predictors, especially when models are applied to novel chemical space. Evidence from recent studies indicates that Evidential Deep Learning provides a strong foundation for efficient and calibrated UQ, striking a balance between performance and computational cost. Further enhancement with Normalizing Flows addresses expressivity limitations of the standard Dirichlet assumption, offering superior robustness in the face of complex data shifts and ambiguous OOD samples.
For researchers and drug development professionals, the choice of method should be guided by the specific application constraints. Standard EDL is suitable for fast, reasonably robust UQ in many practical scenarios. In contrast, Flow-enhanced EDL should be the preferred choice for high-stakes applications where OOD robustness is critical and computational resources allow for its implementation. As the field moves forward, rigorously evaluating models using challenging, cluster-based OOD splits—rather than simpler scaffold splits—will be essential for selecting models that truly generalize to the unknowns of chemical space [16].
For researchers and scientists in drug development, the accuracy of molecular property predictors is paramount. However, a model's true test comes from its out-of-distribution (OOD) robustness—its ability to maintain performance on novel, structurally diverse molecular scaffolds not seen during training. This challenge is frequently compounded by the scarce, incomplete, and imbalanced nature of experimental biochemical datasets [48]. A strategic approach to data, encompassing pre-training, rigorous data splitting, and domain-informed augmentation, is not merely beneficial but essential for building predictive tools that generalize reliably to the chemical space of actual interest, thereby de-risking the early stages of drug discovery.
This guide provides a comparative analysis of data strategies, focusing on their measurable impact on the generalization capabilities of molecular property predictors.
Comparative Analysis of Data Strategies
The quest for robust models has led to several core data strategies. The table below compares their core mechanisms, applications, and proven impacts on generalization.
Table 1: Comparative Analysis of Data Strategies for Generalization
| Strategy | Core Mechanism | Best for Data Scenarios | Impact on OOD Robustness | Key Considerations |
|---|---|---|---|---|
| Pre-training | Leverages knowledge from large, diverse source datasets [49] [50]. | Very small target datasets; large, diverse pre-training data is available [51]. | High effective robustness on out-of-support shifts (extrapolation) [51]. | Data quantity is a key factor; target task alignment improves performance [50]. |
| Strategic Data Splitting | Isoses a "hidden" test set that simulates a realistic OOD evaluation [52]. | All projects, especially those with temporal, sequential, or implicit structural biases. | Prevents over-optimistic performance estimates; is the foundation for true OOD evaluation [52]. | Scaffold-based splitting is crucial in cheminformatics to test generalization to new chemotypes. |
| Data Augmentation | Artificially expands training data using label-preserving transformations [53] [54]. | Small to medium-sized datasets; domains with well-defined invariance and semantic rules. | Improves robustness to intra-distribution variations; can help bridge gaps to specific OOD tests. | Quality and semantic validity of augmented data are critical; domain knowledge is required [54]. |
| Multi-task Learning | Shares representations across related prediction tasks during training [48]. | Multiple related tasks with sparse data; some tasks have more data than others. | Leverages auxiliary tasks to improve generalization and data efficiency on a primary task [48]. | Performance gains depend on the relatedness of the tasks and the sparsity of the primary dataset. |
Quantitative Comparisons in Molecular Property Prediction
Theoretical benefits of these strategies are confirmed by experimental results in molecular property prediction. The following table summarizes findings from controlled studies, providing a performance baseline.
Table 2: Experimental Data on Strategy Performance for Molecular Property Prediction
| Study Focus | Dataset(s) Used | Experimental Setup | Key Quantitative Finding | Implication for Generalization |
|---|---|---|---|---|
| Data Augmentation [54] | Five benchmark molecular datasets | Graph Neural Networks (GNNs) tested with and without topology-based data augmentation. | The proposed augmentation method significantly improved prediction accuracy across tested datasets. | Incorporating domain knowledge (e.g., molecular connectivity indices) into augmentation generates reliable data and improves model accuracy [54]. |
| Multi-task Learning [48] | QM9; real-world sparse fuel ignition dataset | Single-task vs. Multi-task models on progressively larger data subsets. | Multi-task learning outperformed single-task models on small and inherently sparse datasets. | Augmenting a sparse primary dataset with auxiliary tasks, even weakly related ones, enhances predictive accuracy in low-data regimes [48]. |
| Pre-training Data Alignment [50] | Over 10 NLP tasks; scaling laws | 500+ models trained with data selected via benchmark-targeted ranking (BETR). | BETR achieved a 2.1x compute multiplier over baselines, improving performance on 9/10 tasks. | Simply aligning pre-training data with the target task distribution is a highly effective strategy for shaping model capabilities and efficiency [50]. |
Detailed Experimental Protocols
To ensure reproducibility and provide a clear blueprint for implementation, this section details the methodologies behind the key experiments cited.
Protocol: Multi-task Learning for Molecular Property Prediction
This protocol, based on the work of Javaid et al. [48], outlines how to use multi-task learning to mitigate data scarcity.
- Objective: To determine whether a multi-task graph neural network can improve prediction accuracy on a small, sparse primary dataset by leveraging data from auxiliary property prediction tasks.
- Materials & Model: Use a Graph Neural Network (GNN) architecture, such as a Graph Convolutional Network (GCN) or Message Passing Neural Network (MPNN), configured with a shared backbone and task-specific prediction heads.
- Datasets:
- Procedure:
- Data Preprocessing: Standardize all molecular structures (e.g., SMILES strings) and normalize property values per task.
- Model Training:
- Single-Task Baseline: Train the GNN only on the primary dataset. This serves as the performance baseline.
- Multi-Task Model: Jointly train the GNN on the primary dataset and all auxiliary datasets. The loss function is a weighted sum of the losses for each task.
- Evaluation: Evaluate all models on a held-out test set split from the primary dataset. Use performance metrics relevant to the primary task (e.g., Mean Absolute Error for regression).
- Analysis: Compare the performance of the multi-task model against the single-task baseline. A significant improvement in performance on the primary task demonstrates that the auxiliary data provided a beneficial regularization effect, enhancing generalization.
Protocol: Topology-Based Molecular Data Augmentation
This protocol details the method proposed by Wang et al. [54] for generating semantically valid augmented molecular data.
- Objective: To augment a molecular dataset by modifying molecular graphs while preserving their molecular connectivity index, thereby retaining key topology-based physicochemical properties.
- Materials: A dataset of molecular graphs; software to calculate the molecular connectivity index.
- Procedure:
- Calculate Molecular Connectivity Index: For every molecule in the training set, compute its molecular connectivity index (or other relevant topological indices).
- Generate Augmented Data: For each original molecular graph, generate new graph variants by applying topology-modifying operations that are constrained to preserve the calculated connectivity index. These operations could include:
- Bond Rotation: Rotating around single bonds to create different conformers.
- Subgraph Replacement: Swapping substructures with others that have an equivalent topological contribution to the overall index.
- Filtering: Ensure the generated molecules are chemically valid (e.g., correct valences).
- Model Training: Train a GNN on the combined set of original and augmented molecular graphs.
- Analysis: Evaluate the model on a separate, non-augmented test set. Compare its performance against a model trained only on the original data. The use of domain-knowledge-guided augmentation is expected to yield a greater performance boost than naive augmentation methods [54].
Visualizing Data Strategy Workflows
The following diagrams map the logical relationships and workflows of the data strategies discussed, providing a high-level visual guide.
Strategic Roadmap for Data Handling
Pre-training and Fine-tuning for Robustness
The Scientist's Toolkit: Research Reagents & Solutions
Building and evaluating robust molecular property predictors requires a suite of software tools and datasets. The following table catalogs essential "research reagents" for practitioners in the field.
Table 3: Essential Research Reagents for Robust Molecular Predictors
| Tool/Dataset Name | Type | Primary Function | Relevance to Generalization |
|---|---|---|---|
| QM9 Dataset [48] | Dataset | A comprehensive dataset of calculated quantum mechanical properties for small organic molecules. | Serves as a standard benchmark and a valuable source of auxiliary data for multi-task learning and pre-training [48]. |
| Graph Neural Networks (GNNs) | Model Architecture | A class of deep learning models designed to operate directly on graph-structured data, like molecules. | The de facto standard for molecular property prediction, capable of learning from the innate graph structure of molecules. |
| Stratified K-Fold Cross-Validation [53] | Evaluation Protocol | A data resampling technique that ensures proportional representation of classes/values in each fold. | Provides a more reliable estimate of model performance than a single train-test split, especially on imbalanced datasets [53]. |
| Molecular Connectivity Index [54] | Topological Descriptor | A numerical value that summarizes the topology of a molecular graph and correlates with physicochemical properties. | Can guide domain-informed data augmentation by ensuring generated molecules preserve critical topological properties [54]. |
| BETR (Benchmark-Targeted Ranking) [50] | Data Selection Method | A method to select pre-training documents based on similarity to benchmark training examples. | Directly aligns pre-training data with the target task, significantly improving performance and compute efficiency [50]. |
| Scaffold Split | Data Splitting Strategy | A method of splitting a molecular dataset based on the Bemis-Murcko scaffold, grouping molecules that share a core structure. | The gold-standard for simulating a realistic OOD test in drug discovery, evaluating performance on novel molecular scaffolds. |
The generalization of molecular property predictors is not a product of model architecture alone but is fundamentally determined by the data strategy. As the experimental data shows, multi-task learning and domain-informed data augmentation directly address the core problem of data scarcity, yielding measurable improvements in predictive accuracy [48] [54]. Furthermore, the paradigm of pre-training offers a powerful path to robustness, particularly when the pre-training data is aligned with the target task and when the model faces the challenge of extrapolation [50] [51].
However, no strategy is a panacea. The effectiveness of each depends on the specific data context and the nature of the distribution shift. Therefore, a deliberate, combined approach is recommended: using rigorous, domain-aware data splitting for evaluation, enriching training data through informed augmentation or multi-task learning, and leveraging aligned pre-training where feasible. For researchers in drug development, adopting this holistic view of data strategy is a critical step toward building more reliable and impactful predictive models.
Hyperparameter Optimization and Model Selection for Maximum OOD Robustness
The pursuit of accurate molecular property predictors is a central challenge in modern drug discovery and materials science. However, the real-world utility of these models is often determined not by their performance on held-out test data from the same distribution, but by their ability to maintain accuracy when faced with out-of-distribution (OOD) samples—molecules with structural or functional characteristics not adequately represented in the training data. This robustness gap represents a critical bottleneck in the reliable deployment of AI-driven molecular property prediction (MPP) in safety-critical applications.
This guide provides a systematic comparison of hyperparameter optimization (HPO) strategies and model selection techniques specifically evaluated for their ability to enhance OOD robustness in molecular property predictors. By framing HPO not merely as an accuracy-enhancing step but as a crucial component of robustness engineering, we aim to provide researchers with methodologies to develop models that generalize more reliably to novel chemical spaces.
Hyperparameter Optimization Algorithms: A Comparative Analysis for Robustness
Hyperparameter optimization transcends mere accuracy improvement; properly tuned models develop generalized representations that remain stable under distributional shifts. We compare the predominant HPO strategies with a specific focus on characteristics that contribute to OOD robustness.
Table 1: Comparison of Hyperparameter Optimization Methods for OOD Robustness
| Method | Core Mechanism | Computational Efficiency | Robustness Strengths | Key Limitations |
|---|---|---|---|---|
| Grid Search | Exhaustive search over predefined parameter space | Low - Curse of dimensionality | Complete coverage of search space | Impractical for high-dimensional spaces [56] |
| Random Search | Random sampling from parameter distributions | Medium - Better than grid search | Identifies important parameters efficiently [57] | No transfer of knowledge between trials |
| Bayesian Optimization | Probabilistic model guides search | High - Sample-efficient | Intelligent explore/exploit balance [56] | Complex implementation; overhead for model management |
| Hyperband | Adaptive resource allocation + random search | Very High - Early-stopping of poor trials | Rapid identification of promising configurations [57] | Limited guidance on where to sample |
| BOHB (Bayesian Opt + Hyperband) | Bayesian models + early-stopping | Highest - Combines strengths of both | State-of-the-art for complex spaces [57] | Implementation complexity |
| PriMO | Multi-objective BO with expert priors | Varies with prior quality | Explicitly optimizes multiple objectives [58] | New approach; limited community experience |
The choice of HPO algorithm significantly impacts both the final model performance and the computational resources required. For molecular property prediction, where training large neural networks can be computationally intensive, methods that offer early-stopping capabilities like Hyperband and BOHB provide distinct advantages by quickly eliminating unpromising configurations [57]. Bayesian optimization methods excel in sample efficiency but require careful setup of the surrogate model and acquisition function.
For OOD robustness specifically, multi-objective approaches like PriMO (Prior informed Multi-objective Optimizer) show particular promise as they can simultaneously optimize for in-distribution accuracy and robustness metrics, potentially creating models that maintain performance across distributional shifts [58].
Experimental Protocols for Evaluating OOD Robustness
Establishing a Robustness Evaluation Framework
Rigorous evaluation of OOD robustness requires carefully designed experimental protocols that simulate real-world distribution shifts. The following methodology provides a standardized approach for assessing molecular property predictors:
Controlled Data Partitioning: Split datasets using meaningful molecular descriptors (e.g., scaffold-based splits, functional group presence/absence, or physicochemical property ranges) to create systematic distribution shifts rather than random splits [59].
Distance-Based Metric Calculation: Implement quantitative measures of distribution shift using established statistical distances:
- Wasserstein Distance (WD): Effective for high-dimensional data like molecular fingerprints [59]
- Maximum Mean Discrepancy (MMD): Kernel-based method suitable for comparing molecular distributions [59]
- Kolmogorov-Smirnov Statistic (KS): Particularly effective for ImageNet-based models in benchmark studies [59]
Performance Discrepancy Measurement: Calculate robustness metrics including:
- OOD Performance Drop: (ID accuracy - OOD accuracy)
- Relative Performance Ratio: (OOD accuracy / ID accuracy)
- Failure Consistency: Correlation of errors across different OOD splits
HPO Experimental Design for Molecular Property Prediction
Based on recent systematic evaluations, the following protocol ensures comprehensive hyperparameter optimization for deep neural networks in MPP [57]:
Critical Hyperparameter Identification:
- Architectural parameters: number of layers, hidden units, activation functions
- Optimization parameters: learning rate, batch size, optimizer selection
- Regularization parameters: dropout rates, weight decay, early stopping patience
Search Space Definition:
- Learning rate: Log-uniform distribution between 1e-5 and 1e-2
- Hidden units: Integer values between 64 and 1024
- Dropout rate: Uniform distribution between 0.0 and 0.5
- Batch size: Categorical values from {32, 64, 128, 256}
Evaluation Protocol:
- Use nested cross-validation with independent OOD test sets
- Implement multiple random seeds to account for optimization instability
- Track both convergence speed and final performance
Robustness Evaluation Workflow: This diagram illustrates the comprehensive process for evaluating model robustness, from data preparation through hyperparameter optimization to final model selection.
Advanced HPO Techniques for Enhanced Robustness
Multi-Objective Optimization with PriMO
The PriMO algorithm represents a significant advancement for OOD robustness applications by enabling simultaneous optimization of multiple competing objectives [58]. Unlike single-objective HPO that focuses solely on accuracy, PriMO can explicitly balance:
- Primary Objective: Prediction accuracy on validation data
- Robustness Objectives: Performance consistency across distribution shifts
- Efficiency Objectives: Inference speed, memory footprint, computational requirements
The algorithm incorporates expert priors about potentially robust configurations, accelerating the discovery of models that maintain performance under distribution shifts. This is particularly valuable in molecular property prediction where domain knowledge about molecular representations exists.
Automated Model Selection with Meta-Learning
For complex deployment environments with multiple potential OOD scenarios, meta-learning approaches like M3OOD provide automated model selection capabilities [60]. This framework:
- Learns from historical model behaviors across diverse distribution shifts
- Combines multimodal embeddings with handcrafted meta-features
- Recommends suitable detectors for new data distribution shifts with minimal supervision
- Has demonstrated consistent outperformance over static selection baselines across diverse test scenarios [60]
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2: Essential Tools for Robust Molecular Property Prediction Research
| Tool/Category | Specific Examples | Function in Robustness Research |
|---|---|---|
| HPO Frameworks | Optuna, KerasTuner, Ray Tune | Automated hyperparameter search with advanced algorithms like Bayesian optimization and Hyperband [57] [56] |
| Robustness Metrics | Wasserstein Distance, MMD, KS statistic | Quantify distribution shift and model robustness [59] |
| Molecular ML Libraries | DeepChem, DGLLifeSci, MAT | Specialized architectures for molecular graph processing |
| Model Architectures | GNNs, KA-GNNs, Transformers | Advanced architectures with built-in robustness characteristics [61] |
| OOD Detection | M3OOD framework | Automated selection of appropriate OOD detectors for new distribution shifts [60] |
| Visualization Tools | RDKit, ChemPlot | Analysis of chemical space coverage and identification of distribution gaps |
Comparative Performance Analysis
Empirical Results Across Molecular Benchmarks
Recent systematic evaluations provide quantitative insights into HPO method performance for molecular property prediction:
Table 3: Performance Comparison of HPO Methods on Molecular Property Prediction Tasks
| HPO Method | Average Accuracy Gain vs Default | Computational Efficiency | OOD Robustness | Implementation Complexity |
|---|---|---|---|---|
| Random Search | 7-12% | Medium | Variable | Low [57] |
| Bayesian Optimization | 10-15% | High (sample-efficient) | Good with proper metrics | Medium [57] |
| Hyperband | 12-16% | Very High | Consistent | Low-Medium [57] |
| BOHB | 14-18% | Highest | Strong | High [57] |
These results demonstrate that while all HPO methods provide significant improvements over default hyperparameters, more advanced methods like BOHB offer the best balance of performance and efficiency. The critical finding for robustness-focused applications is that proper HPO consistently enhances OOD performance even when optimization is conducted solely on in-distribution data, suggesting that well-tuned models develop more generalized representations.
Architectural Innovations for Enhanced Robustness
Beyond conventional HPO, architectural innovations can significantly impact OOD robustness. Recent advances like Kolmogorov-Arnold Graph Neural Networks (KA-GNNs) demonstrate how fundamental architectural changes can enhance both accuracy and robustness [61]:
- KA-GNNs integrate Fourier-based KAN modules into all core GNN components (node embedding, message passing, readout)
- Theoretical advantages include stronger approximation capabilities and smoother gradients
- Empirical results show consistent outperformance of conventional GNNs across multiple molecular benchmarks
- Interpretability benefits enable identification of chemically meaningful substructures that contribute to predictions
These architectural advancements, when combined with rigorous HPO, create molecular property predictors with significantly enhanced OOD robustness profiles.
Integrated Workflow for Maximum Robustness
Integrated Robustness Optimization Pipeline: A comprehensive workflow combining multi-objective HPO with rigorous validation and deployment protocols for maximum OOD robustness.
Achieving maximum OOD robustness in molecular property predictors requires moving beyond conventional hyperparameter optimization approaches focused solely on accuracy maximization. The experimental evidence and comparative analysis presented in this guide demonstrate that:
- Advanced HPO methods like BOHB and PriMO significantly outperform simpler approaches in both efficiency and final robustness
- Multi-objective optimization frameworks that explicitly balance accuracy with robustness metrics produce models with superior OOD performance
- Architectural innovations like KA-GNNs provide fundamental advantages for learning robust molecular representations
- Systematic evaluation protocols using appropriate distance metrics and OOD scenarios are essential for reliable robustness assessment
For researchers and practitioners in drug discovery and materials science, adopting these advanced HPO and model selection strategies can dramatically improve the real-world reliability of molecular property predictors, accelerating the translation of computational models into practical scientific and commercial applications.
Benchmarking Performance: A Rigorous Framework for Validating OOD Robustness
The accelerating integration of machine learning (ML) into molecular discovery has created a pressing need for models that perform reliably in real-world scenarios. A significant frontier challenge in this domain is out-of-distribution (OOD) generalization—the ability of models to make accurate predictions on molecules that extend beyond the chemical space or property ranges seen during training [9]. The inherent goal of molecular discovery is to identify novel compounds with exceptional properties, a task that is fundamentally OOD. Despite its importance, a comprehensive understanding of model performance under these conditions has been lacking. The BOOM (Benchmarking Out-Of-distribution Molecular property predictions) study addresses this gap by conducting a large-scale, systematic evaluation of over 140 combinations of models and property prediction tasks to benchmark their OOD performance [9]. This work establishes that achieving strong OOD generalization is a pivotal challenge for the future of chemical ML, as even top-performing models exhibit a substantial performance drop when applied to OOD data [9].
BOOM Benchmark Design and Methodology
Datasets and OOD Splitting Strategy
The BOOM benchmark is constructed using ten distinct molecular property datasets to ensure diversity and comprehensiveness [9]. Eight properties are sourced from the widely used QM9 dataset, which contains Density Functional Theory (DFT) calculations for approximately 133,886 small organic molecules (CHONF atoms). These properties include isotropic polarizability (α), heat capacity (Cv), HOMO energy, LUMO energy, HOMO-LUMO gap, dipole moment (μ), electronic spatial extent (R²), and zero-point vibrational energy (zpve) [9]. Additionally, two properties—density and solid heat of formation—are taken from the 10k Dataset, which is derived from 10,206 experimentally synthesized CHON molecules from the Cambridge Crystal Structure Dataset [9].
A critical aspect of the benchmark is its methodology for defining and creating OOD splits. Instead of partitioning data based on input chemical structures, BOOM adopts a property-based OOD splitting approach, which aligns directly with the objectives of molecule discovery [9]. For each molecular property, the methodology involves:
- Fitting a Kernel Density Estimator: A Gaussian kernel density estimator is applied to the distribution of property values for a given dataset [9].
- Calculating Probability Scores: The probability of each molecule, given its property value, is computed using this estimator [9].
- Selecting OOD Splits: Molecules with the lowest probability scores are assigned to the OOD test set. This typically corresponds to molecules at the tail ends of the property value distribution. In QM9, the lowest 10% of probability scores form the OOD set, while for the 10k dataset, the lowest 1000 molecules are selected [9].
- Creating ID and Training Splits: The remaining molecules are randomly sampled to create an in-distribution (ID) test set (10% for QM9, 5% for 10k), with the rest allocated for training and validation [9].
This strategy ensures the OOD benchmark evaluates a model's capability to extrapolate to property values not represented in the training data, which is essential for discovering high-performance materials and molecules [9].
Evaluated Models and Architectures
The benchmark encompasses a wide array of ML models, ranging from traditional approaches to advanced deep learning architectures, providing a holistic comparison landscape [9]. The evaluated models can be categorized as follows:
- Molecule Featurizer-based Models: These models use engineered molecular features as input. The benchmark includes Random Forest coupled with RDKit Molecular Descriptors (125 chemical features and 86 functional group features) as a baseline [9].
- Transformer Models: Several transformer-based architectures, pre-trained on large chemical corpora, were evaluated:
- ChemBERTa: An encoder-only model with a BERT architecture trained on PubChem [9].
- MolFormer: An encoder-decoder model with a T5 backbone trained on PubChem [9].
- Regression Transformer (RT): An XLNet-based model capable of masked language modeling and autoregressive generation [9].
- ModernBERT: A state-of-the-art encoder-only model incorporating rotary positional embeddings and other architectural improvements [9].
- Graph Neural Networks (GNNs): Multiple GNN architectures with different representational biases were included:
- Chemprop and TGNN: Standard message-passing GNNs operating on atom and bond features, with permutation invariance [9].
- IGNN: An E(3)-invariant GNN that incorporates pair-wise atomic distances [9].
- EGNN: An E(3)-equivariant GNN that explicitly models atom positions [9].
- MACE: A model that also uses pair-wise distances and angles [9].
Table 1: Overview of Model Architectures Evaluated in the BOOM Benchmark
| Model Name | Architecture Type | Molecular Representation | Key Invariance/Equivariance | Parameter Count |
|---|---|---|---|---|
| Random Forest | Ensemble | RDKit Descriptors | N/A | N/A |
| ChemBERTa | Transformer | SMILES | N/A | 83 Million |
| MolFormer | Transformer | SMILES | N/A | 48 Million |
| RT | Transformer | SMILES | N/A | 27 Million |
| ModernBERT | Transformer | SMILES | N/A | 111 Million |
| Chemprop | GNN | Graph (Atoms, Bonds) | Permutation | ~200,000 |
| TGNN | GNN | Graph (Atoms, Bonds) | Permutation | ~200,000 |
| IGNN | GNN | Graph + Distances | E(3)-Invariant | ~217,000 |
| EGNN | GNN | Graph + Positions | E(3)-Equivariant | ~217,000 |
| MACE | GNN | Graph + Distances/Angles | E(3)-Invariant | Information Missing |
Key Experimental Findings and Performance Analysis
The large-scale evaluation across 10 OOD tasks and 12 ML models yielded a sobering conclusion: no single existing model demonstrates strong OOD generalization across all tasks [9]. This finding underscores the pervasive difficulty of the OOD problem in molecular property prediction. A particularly telling result is that even the top-performing model in the benchmark suffered from an average OOD error that was three times larger than its in-distribution error [9]. This performance gap highlights the substantial risk of relying solely on ID metrics for model selection in discovery-oriented applications.
Further analysis revealed that the relationship between ID and OOD performance is not always straightforward. While a strong positive correlation (Pearson r ~ 0.9) exists between ID and OOD performance for simpler splitting strategies like scaffold splitting, this correlation significantly weakens (Pearson r ~ 0.4) for more challenging, cluster-based OOD splits [16]. This suggests that a model excelling on ID data cannot be automatically assumed to perform well on all types of OOD data, emphasizing the need for targeted OOD evaluation based on the intended application domain [16].
Impact of Model Architecture and Training Strategies
The benchmark provides detailed insights into how architectural choices and training strategies influence OOD generalization:
- Inductive Biases in GNNs: Models with high, physically motivated inductive biases, such as E(3)-invariant and E(3)-equivariant GNNs, demonstrated strong performance on OOD tasks involving simple, specific properties where these biases are relevant [9].
- Chemical Foundation Models: Transformer-based models, pre-trained on large datasets like PubChem, offer promise in data-scarce scenarios through transfer learning [9]. However, the benchmark results indicate that current chemical foundation models do not yet exhibit strong OOD extrapolation capabilities [9]. Their performance did not consistently surpass that of simpler, task-specific models on the OOD tasks.
- Ablation Studies: Extensive ablation experiments within the BOOM study highlighted that factors such as data generation procedures, pre-training strategies, hyperparameter optimization, and molecular representations all significantly impact OOD performance [9].
Alternative Approaches for OOD Prediction
Complementary to the BOOM benchmark, other research avenues are being explored to address the OOD challenge:
- Bilinear Transduction: This transductive method re-frames the prediction problem. Instead of predicting a property for a new material directly, it learns how property values change as a function of differences in material representation space [2]. During inference, predictions are made based on a chosen training example and the representation-space difference between it and the new sample [2]. This approach has shown promising results, improving extrapolative precision by 1.8× for materials and 1.5× for molecules, and boosting the recall of high-performing candidates by up to 3× [2].
- Data Densification: Another proposed technique addresses covariate shift and data scarcity by leveraging unlabeled data to create interpolations between ID and OOD samples. A bilevel optimization framework learns how to generalize beyond the training distribution, demonstrating performance gains on real-world datasets with substantial distribution shifts [62].
Experimental Protocols and Workflows
BOOM Benchmarking Workflow
The following diagram illustrates the end-to-end workflow for the BOOM benchmarking methodology, from dataset preparation to model evaluation.
Transductive Prediction via Bilinear Transduction
The Bilinear Transduction method, which showed improved OOD extrapolation, follows a distinct workflow centered on analogical reasoning.
To facilitate the reproduction and extension of these benchmarking efforts, the following table details essential computational "reagents" and resources.
Table 2: Essential Research Reagents and Resources for OOD Benchmarking
| Category | Item/Resource | Description | Function in Research | Source/Availability |
|---|---|---|---|---|
| Datasets | QM9 Dataset | ~134k small organic molecules with DFT-calculated quantum chemical properties. | Primary benchmark for quantum properties; provides standardized comparison base. | Publicly Available |
| 10k Dataset | ~10k experimentally synthesized crystals from the CSD with DFT properties (density, Hf). | Benchmark for solid-state properties with experimental structures. | Publicly Available | |
| MoleculeNet | Curated collection of molecular datasets for various property prediction tasks. | Source of diverse benchmarks (e.g., ESOL, FreeSolv) for OOD evaluation. | Publicly Available | |
| Software & Models | BOOM Benchmark | Standardized benchmark suite for OOD molecular property prediction. | Provides evaluation framework, splitting methods, and baseline results for comparison. | GitHub (LLNL) |
| MatEx (Materials Extrapolation) | Implementation of the Bilinear Transduction method for OOD prediction. | Enables transductive learning experiments for improved extrapolation. | GitHub (learningmatter-mit) | |
| RDKit | Open-source cheminformatics toolkit. | Used for molecule handling, descriptor calculation, and fingerprint generation. | Publicly Available | |
| DeepChem | Open-source toolkit for deep learning in drug discovery, materials science, and quantum chemistry. | Provides implementations of various molecular ML models and utilities. | Publicly Available | |
| Molecular Representations | RDKit Descriptors | 211 chemically-informed features (molecular weight, functional groups, etc.). | Input for traditional machine learning models (e.g., Random Forest). | Via RDKit |
| SMILES String | Text-based representation of molecular structure. | Input for Transformer-based models (e.g., ChemBERTa, MolFormer). | Standard | |
| Molecular Graph | Graph with atoms as nodes and bonds as edges. | Native input for Graph Neural Networks (GNNs) like Chemprop, EGNN. | Standard |
The comprehensive benchmarking of over 140 model-task combinations establishes that out-of-distribution generalization remains a significant, unsolved challenge in molecular property prediction. The BOOM benchmark provides a crucial foundation for the community, revealing that no current model consistently excels across all OOD tasks and that architectural choices, pre-training strategies, and representation learning all profoundly impact OOD performance [9]. Promising paths forward include the development of models with stronger physical inductive biases, innovative training paradigms like transductive learning [2] and data densification [62], and the continued expansion of robust benchmarking standards. For researchers and drug development professionals, these findings underscore the critical importance of validating models against OOD metrics that reflect real-world discovery goals, moving beyond the potentially misleading comfort of in-distribution performance. The pursuit of ML models with true OOD robustness is now a defining frontier for the field of AI-driven molecular discovery [9].
The ability of machine learning (ML) models to generalize to out-of-distribution (OOD) data is a critical frontier in computational chemistry and materials science. Molecular discovery is inherently an OOD prediction problem, as identifying novel compounds requires extrapolating beyond the boundaries of known chemical space or property values [9]. This guide provides a comparative analysis of performance metrics—specifically extrapolative precision, recall, and Mean Absolute Error (MAE)—for evaluating the OOD robustness of molecular property predictors. We synthesize findings from recent benchmark studies to objectively assess the current state of the field and provide researchers with standardized methodologies for rigorous model evaluation.
Key Performance Metrics for OOD Evaluation
Evaluating models on OOD data requires a distinct set of metrics that capture different aspects of extrapolative performance. The most relevant metrics for molecular property prediction include:
- Extrapolative Precision: Measures the fraction of true high-performing candidates among those identified by the model. This is crucial for virtual screening campaigns where resource efficiency depends on minimizing false positives [2].
- Recall: Quantifies the model's ability to identify all true high-performing candidates from the OOD set. High recall ensures that promising candidates are not overlooked during screening [2].
- Mean Absolute Error (MAE): A standard regression metric that measures the average magnitude of errors between predicted and actual property values, providing insight into the general accuracy of predictions in OOD settings [2] [63].
- OOD/ID Error Ratio: Compares the MAE on OOD data to the MAE on in-distribution data, with a higher ratio indicating greater performance degradation when extrapolating [9].
Comparative Performance Analysis of Molecular Property Predictors
Recent large-scale benchmarking efforts have revealed significant variations in how different model architectures handle OOD data. The table below summarizes the OOD performance of various approaches across multiple molecular property prediction tasks.
Table 1: OOD Performance of Molecular Property Prediction Models
| Model Category | Model Name | Key OOD Findings | Extrapolative Performance | Recommended Use Cases |
|---|---|---|---|---|
| Classical ML | Random Forest (RDKit) | Moderate OOD degradation; performance varies by splitting strategy [64] | MAE increase of 1.5-2× over ID | Baseline comparisons; scaffold-based splits |
| Graph Neural Networks | Chemprop | Struggles with complex OOD tasks; average OOD error 3× larger than ID [9] | Low extrapolative precision on cluster splits | ID tasks with simple OOD requirements |
| Transformer Models | ChemBERTa | Limited OOD extrapolation despite pre-training [9] | Inconsistent across property types | Transfer learning on similar chemical spaces |
| Physically-Informed Models | EGNN | Better OOD generalization for geometry-sensitive properties [9] | Improved recall on tail distributions | Quantum mechanical properties |
| Specialized OOD Methods | Bilinear Transduction | Improves extrapolation to high-value property ranges [2] | 1.5× higher precision; 3× higher recall | Virtual screening for extreme properties |
The BOOM benchmark (Benchmarking Out-Of-distribution Molecular property predictions), which evaluated over 140 model-task combinations, found that no existing model achieves strong OOD generalization across all tasks [9]. Even top-performing models exhibited an average OOD error approximately three times larger than their in-distribution error. This performance gap highlights the significant challenge of OOD generalization in molecular property prediction.
Experimental Protocols for OOD Evaluation
OOD Data Splitting Strategies
The methodology for creating OOD splits significantly impacts benchmark results and model evaluation.
Table 2: OOD Data Splitting Methodologies
| Splitting Method | Description | OOD Challenge Level | ID-OOD Performance Correlation |
|---|---|---|---|
| Random Splitting | Standard random partition of dataset | Low (baseline) | Strong (r ~0.9) [64] |
| Scaffold Splitting | Groups molecules by Bemis-Murcko scaffolds | Moderate | Strong (r ~0.9) [16] [64] |
| Cluster-Based Splitting | Uses chemical similarity clustering (ECFP4 fingerprints) | High | Weak (r ~0.4) [16] [64] |
| Property Value Splitting | Holds out tail ends of property value distribution [9] | Variable | Depends on property |
| Element-Based Splitting | Holds out specific elements from training [63] | High for composition-based models | Typically weak |
Research shows that cluster-based splitting using chemical similarity poses the most significant challenge for both classical ML and graph neural network models, resulting in the weakest correlation between ID and OOD performance [16] [64]. This makes it particularly valuable for stress-testing model robustness.
Benchmarking Workflow
The following diagram illustrates a standardized experimental workflow for OOD evaluation of molecular property predictors:
Relationship Between Model Capabilities and OOD Performance
The OOD performance of molecular property predictors is influenced by multiple architectural and methodological factors. The following diagram maps these key relationships:
Key insights from recent studies include:
- Physical encoding of atomic information (using properties like electronegativity, atomic radius) significantly improves OOD performance compared to one-hot encoding, particularly for small datasets [63].
- Model architecture alone doesn't guarantee OOD robustness; current transformer-based foundation models show limited OOD extrapolation capabilities despite extensive pre-training [9].
- Training strategies such as transfer learning and fine-tuning have variable effects on OOD performance, with outcomes highly dependent on the similarity between pre-training and target domains [9].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Resources for OOD Molecular Property Prediction Research
| Resource Category | Specific Tools | Function in OOD Research |
|---|---|---|
| Benchmark Datasets | QM9 [9], TDC [64], Matbench [2] | Standardized datasets for reproducible OOD evaluation across diverse chemical properties |
| OOD Splitting Tools | BOOM [9], Cluster-based Splitters [16] | Methodologies for creating meaningful OOD test sets that challenge model generalization |
| Molecular Representations | RDKit Descriptors [9], ECFP4 Fingerprints [64], SMILES [9] | Converting molecular structures into model-input features with varying OOD robustness |
| Model Architectures | GNNs (Chemprop) [9], Transformers (ChemBERTa) [9], Random Forests [64] | Diverse modeling approaches with different OOD generalization capabilities |
| Evaluation Metrics | Extrapolative Precision [2], OOD/ID Error Ratio [9], MAE [2] | Quantifying different aspects of OOD performance for comprehensive assessment |
| Physical Encoding | CGCNN encoding [63], MEGNet encoding [63] | Incorporating domain knowledge to improve model generalization beyond training distribution |
The systematic evaluation of extrapolative precision, recall, and MAE on OOD data reveals significant challenges in molecular property prediction. Current state-of-the-art models, including graph neural networks and transformer-based approaches, struggle with consistent OOD generalization, particularly under challenging splitting strategies like cluster-based division of chemical space. The research community has responded with specialized benchmarks like BOOM and methodologies like bilinear transduction that show promise for improving extrapolation to high-value property ranges. For researchers and drug development professionals, selecting models based on comprehensive OOD evaluation—rather than in-distribution performance alone—is crucial for deploying reliable predictors in real-world discovery pipelines. The continued development of standardized benchmarks, physically-informed model architectures, and specialized OOD evaluation metrics will be essential for advancing robust molecular property prediction.
The accurate prediction of molecular properties, including bioactivity and Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) profiles, is a critical challenge in modern drug discovery. The high failure rates of drug candidates due to unfavorable properties have intensified the search for robust computational prediction models [65]. This guide provides a comparative analysis of three predominant computational approaches: Classical Machine Learning (ML), Graph Neural Networks (GNNs), and Transformer-based models, with a specific focus on their performance and out-of-distribution (OOD) robustness—a key requirement for real-world deployment where models encounter molecules structurally distinct from their training data [66].
Core Technologies and Molecular Representations
The performance of predictive models in computational chemistry is fundamentally linked to how molecules are represented digitally. The three classes of models compared here leverage fundamentally different representation paradigms.
Classical ML Models: These models rely on handcrafted molecular representations. Key examples include:
- Molecular Fingerprints (e.g., Morgan/ECFP): Bit vectors that indicate the presence or absence of specific substructures or topological patterns within the molecule [67] [65].
- RDKit 2D Descriptors: A set of predefined numerical values quantifying physicochemical properties (e.g., molecular weight, logP, polar surface area) [68] [67].
- These fixed representations are then used as input for algorithms such as Random Forest (RF), Support Vector Machines (SVM), and gradient-boosting frameworks like XGBoost and LightGBM [68] [67].
Graph Neural Networks (GNNs): GNNs, including Message Passing Neural Networks (MPNNs), represent a molecule natively as a graph where atoms are nodes and bonds are edges [69] [70]. Through a "message-passing" mechanism, each atom iteratively aggregates information from its neighboring atoms. This process creates learned numerical representations (embeddings) that capture both the local atomic environment and the overall molecular structure in an end-to-end fashion, without relying on pre-defined features [67] [65].
Transformer Models: Originally designed for natural language processing, Transformers have been adapted to chemistry by treating molecular structures as sequential data (e.g., SMILES strings) or sets of fragments [71] [65]. Their core mechanism, self-attention, allows the model to weigh and contextualize the importance of every part of the input sequence relative to all others. This enables them to capture complex, long-range dependencies within a molecule that are often challenging for GNNs, which are more focused on local connectivity [65]. Specialized architectures like MSformer-ADMET further innovate by representing molecules as collections of chemically meaningful "meta-structure" fragments, which are then processed by the Transformer encoder [65].
Performance Benchmarking on Standard Tasks
Numerous studies have systematically evaluated these model classes across various ADMET and bioactivity prediction tasks. The results indicate that the optimal model choice is often task-dependent, but general trends are emerging.
Table 1: Comparative Performance of Model Architectures on ADMET Tasks
| Model Category | Example Algorithms | Key Strengths | Reported Performance (Dataset Example) | Key Limitations |
|---|---|---|---|---|
| Classical ML | XGBoost, Random Forest, SVM [68] [67] | High interpretability, computational efficiency, performs well with small datasets [68] | Best predictor for Caco-2 permeability (XGBoost) [68]; Competitive on various ADMET tasks [67] | Limited ability to generalize beyond chemical space of handcrafted features |
| Graph Neural Networks (GNNs) | MPNN (e.g., Chemprop), GCN [67] [65] | Learns features directly from molecular structure; strong on local structure-property relationships [69] [70] | State-of-the-art on many bioactivity tasks [66]; Strong performance in multi-task learning [65] | Struggles with long-range dependencies; message-passing can lead to over-smoothing [65] |
| Transformers | MSformer-ADMET, BioBERT, Molecule Transformer [71] [65] | Excels at capturing long-range dependencies; flexible pre-training on large unlabeled corpora [65] | Superior performance across 22 ADMET tasks in TDC [65]; Effective in biomedical NLP tasks [71] | High computational cost; requires large datasets for effective training [65] |
The table above summarizes the general characteristics of each model class. A more detailed benchmarking study across 14 different machine learning models, including classical approaches and GNNs, on eight molecular property datasets revealed that the best-performing model is often dataset-specific [66]. However, Tree-based methods (e.g., Random Forest) and Message-Passing Neural Networks (MPNNs) frequently emerge as top performers on many tasks [67] [66]. For instance, in predicting Caco-2 permeability for intestinal absorption, XGBoost demonstrated superior performance on test sets compared to several other models, including Random Forest, SVM, and deep learning models like DMPNN [68].
Meanwhile, advanced Transformer architectures are showing remarkable results. The MSformer-ADMET model, which uses a fragment-based molecular representation, consistently outperformed conventional SMILES-based and graph-based models across a wide range of 22 ADMET endpoints from the Therapeutics Data Commons (TDC) [65].
Critical Analysis: Out-of-Distribution (OOD) Robustness
A model's performance on data that comes from the same distribution as its training set (in-distribution, or ID) can be misleading. Real-world drug discovery often involves projecting into novel chemical spaces, making a model's robustness on OOD data a critical metric.
Defining OOD Splits and Model Performance
Research indicates that the strategy used to split data for OOD testing significantly impacts the observed performance gap between model classes [66].
Table 2: Impact of Data Splitting Strategies on OOD Robustness
| Splitting Strategy | Description | Impact on Model Performance | Correlation between ID and OOD Performance |
|---|---|---|---|
| Random Split | Compounds randomly assigned to train/test sets. | Minimal performance drop. Not a rigorous test of OOD robustness. | Strongly positive (not representative of real-world challenges) |
| Scaffold Split | Train and test sets contain distinct molecular scaffolds (core structures). | Performance drops are moderate for both classical ML and GNNs. Does not pose the greatest challenge [66]. | Strong (Pearson's r ~ 0.9) [66] |
| Cluster-Based Split (UMAP + ECFP4) | Clusters based on chemical similarity; entire clusters held out for testing. | Presents the most challenging scenario. Significant performance drop for all models [66]. | Weak (Pearson's r ~ 0.4) [66] |
A key finding is that both classical ML and GNN models generalize surprisingly well under scaffold splits, with performance "not substantially different from random splitting" [66]. The true test of robustness comes from more rigorous, cluster-based splits, which better simulate the real-world scenario of evaluating truly novel chemotypes.
Relationship Between In-Distribution and OOD Performance
The strength of the correlation between a model's ID performance and its OOD performance is heavily influenced by the splitting strategy, as noted in Table 2. Under scaffold splits, this correlation is strong (Pearson's r ~ 0.9), meaning selecting the best-performing ID model generally guarantees the best OOD performance. However, under the more challenging cluster-based splits, this correlation weakens significantly (Pearson's r ~ 0.4). This suggests that in rigorous OOD settings, model selection based solely on ID performance is unreliable and must be replaced with evaluation protocols that directly assess OOD robustness [66].
Experimental Protocols for Robust Benchmarking
To ensure fair and meaningful comparisons, the following experimental protocols, synthesized from recent rigorous studies, are recommended.
Data Curation and Cleaning
Inconsistent and noisy data are major obstacles in molecular property prediction. A robust data cleaning pipeline is essential [67]:
- Standardization: Use tools like the
MolStandardizemodule from RDKit to generate consistent SMILES representations, adjust tautomers, and handle neutralization [68] [67]. - Salt Removal: Strip salt and counterions from parent organic compounds to focus on the active molecule's properties [67].
- Deduplication: Remove duplicate molecules, keeping the first entry if target values are consistent, or removing the entire group if values are inconsistent [67].
- Visual Inspection: For smaller datasets, use tools like DataWarrior for final manual inspection [67].
Model Training and Evaluation
- Splitting Strategies: Implement multiple data-splitting strategies, including random, scaffold, and cluster-based splits, to thoroughly evaluate both ID and OOD performance [66].
- Hyperparameter Optimization: Conduct extensive hyperparameter tuning for all models to ensure fair comparisons. Studies that skip this step may report biased results [67].
- Statistical Validation: Employ cross-validation coupled with statistical hypothesis testing (e.g., paired t-tests) to determine if performance differences between models are statistically significant [67].
- External Validation: Whenever possible, validate models trained on public data against proprietary in-house datasets from pharmaceutical companies to assess real-world transferability [68].
The following diagram illustrates a standardized workflow for a robust comparative analysis integrating these protocols.
Successful implementation of molecular property prediction models relies on a suite of software tools and data resources.
Table 3: Essential Research Reagents for Molecular Property Prediction
| Resource Name | Type | Primary Function | Relevance to Model Classes |
|---|---|---|---|
| RDKit [68] [67] | Cheminformatics Library | Generation of molecular descriptors (RDKit 2D), fingerprints (Morgan), and molecule standardization. | Core for Classical ML; Preprocessing for GNNs/Transformers |
| Therapeutics Data Commons (TDC) [67] [65] | Data Repository | Curated benchmark datasets for ADMET and bioactivity prediction. | Standardized evaluation for all model classes |
| Chemprop [68] [67] | Software Framework | Implementation of Message Passing Neural Networks (MPNNs) for molecular property prediction. | Primary tool for GNN development and benchmarking |
| DeepChem [67] | Deep Learning Library | Provides a variety of deep learning models and tools for drug discovery. | Training and evaluation for GNNs and other deep models |
| scikit-learn | ML Library | Implementations of classical ML algorithms (Random Forest, SVM, etc.). | Core for Classical ML models |
| XGBoost / LightGBM [68] [67] | Software Library | Efficient implementations of gradient boosting algorithms. | Key for high-performing Classical ML models |
| Transformers Library (Hugging Face) [71] | Software Library | Repository and framework for pre-trained Transformer models. | Adaptation of language models to molecular data |
The comparative analysis reveals that no single model class is universally superior for all ADMET and bioactivity tasks. Classical ML models, particularly XGBoost and Random Forest, remain strong, interpretable, and data-efficient contenders, especially on smaller datasets. GNNs excel at learning directly from molecular structure and have set new standards on many benchmarks. Transformers, with their ability to capture long-range dependencies and their power from large-scale pre-training, are emerging as front-runners, particularly in complex, multi-task ADMET prediction scenarios.
However, the critical differentiator for practical application is out-of-distribution robustness. Evaluations must move beyond simple random or scaffold splits and incorporate more realistic, challenging data partitioning methods like cluster-based splits. The weak correlation between ID and OOD performance under these conditions necessitates a shift in model selection paradigms. Future work should focus on developing models and training strategies explicitly designed for OOD generalization, such as advanced data augmentation, transfer learning, and self-supervised pre-training on diverse chemical spaces, to build predictive tools that truly deliver in the novel chemical frontiers of drug discovery.
In the high-stakes field of computational drug discovery, the ability of machine learning (ML) models to accurately predict molecular properties for novel, out-of-distribution (OOD) compounds is paramount. While models often demonstrate exceptional in-distribution (ID) performance, this proficiency frequently fails to translate to real-world discovery scenarios where models encounter chemically distinct structures. This article presents a comparative analysis of molecular property predictors, examining the critical relationship between ID performance and OOD success. Through systematic evaluation of experimental data and methodologies, we provide researchers and drug development professionals with a framework for assessing model robustness, ultimately guiding the selection of predictive tools capable of accelerating reliable molecular discovery.
Comparative Performance Analysis of Molecular Property Predictors
Table 1: Summary of Model Performance on OOD Molecular Property Prediction Tasks (adapted from BOOM Benchmark [9])
| Model Category | Example Models | Avg. ID Performance (MAE) | Avg. OOD Performance (MAE) | OOD/ID Error Ratio | Key Strengths | Key Limitations |
|---|---|---|---|---|---|---|
| Traditional ML | Random Forest (RDKit) | Varies by dataset | Varies by dataset | ~3x (Average across top models) | Computationally efficient; good baselines | Limited extrapolation capability |
| Graph Neural Networks | Chemprop, TGNN, IGNN, EGNN, MACE | Varies by dataset | Varies by dataset | ~3x (Average across top models) | High inductive bias; effective on specific OOD tasks with simple properties | Inconsistent performance across diverse OOD tasks |
| Transformer Models | ChemBERTa, MolFormer, Regression Transformer, ModernBERT | Varies by dataset | Varies by dataset | ~3x (Average across top models) | Transfer and in-context learning; promising for data-limited scenarios | Current models show weak OOD extrapolation |
The benchmarking data reveals a consistent generalization gap. Even top-performing models exhibit an average OOD error approximately three times larger than their ID error [9]. This indicates that high ID accuracy is not a reliable indicator of OOD success. No single model architecture currently achieves strong OOD generalization across all chemical tasks, establishing this as a frontier challenge in the field [9].
Performance varies significantly based on the type of distribution shift. Models may generalize well to new elemental compositions but fail dramatically on structurally novel scaffolds. For instance, in leave-one-element-out tasks, models show surprisingly robust performance for most elements but display systematic biases and poor R² scores for specific nonmetals like Hydrogen (H), Oxygen (O), and Fluorine (F) [24].
Experimental Protocols for OOD Evaluation
OOD Splitting Strategies
A critical methodological component is the strategy for partitioning data into ID and OOD sets. The BOOM benchmark employs a property-based splitting approach, defining OOD as a "complement distribution with respect to the targets" [9]. The protocol involves:
- Probability-Based Splitting: A kernel density estimator (with Gaussian kernel) is fitted to the property values of the entire dataset.
- OOD Set Selection: Molecules with the lowest probability scores (e.g., the lowest 10% for QM9 dataset) are assigned to the OOD test set. This captures molecules at the tail ends of the property value distribution.
- ID Set Construction: The remaining molecules are randomly sampled to create an ID test set and a training/validation set [9].
Alternative splitting strategies include heuristic-based splits grounded in chemical knowledge, such as:
- Leave-one-X-out: Holding out all molecules containing a specific element, belonging to a certain period/group, or possessing a specific crystal system or space group [24].
- Scaffold-based Splits: Separating molecules based on fundamental molecular frameworks to assess generalization to novel chemotypes.
The BOOM Evaluation Workflow
The following diagram illustrates the standard workflow for benchmarking OOD generalization, as implemented in benchmarks like BOOM.
Key Challenges in OOD Generalization
The Interpolation vs. Extrapolation Dilemma
A pivotal finding from recent studies is that many heuristic-based OOD tasks do not constitute true extrapolation [24]. Analysis of the materials representation space shows that test data from many "OOD" tasks actually reside within regions well-covered by the training data. This leads to an overestimation of model generalizability and the purported benefits of model scaling [24]. Genuinely challenging OOD tasks involve test data that falls outside the training domain, where scaling up training set size or training time yields only marginal improvement or even performance degradation [24].
The Impact of Spurious Correlations
Models often exploit spurious correlations between non-causal (nuisance) features and labels present in the training data. This leads to failures on OOD inputs that share the same nuisance features (e.g., common molecular backgrounds or substructures) but have different semantic labels (e.g., a new protein target) [72] [73]. The strength of this spurious correlation directly impacts OOD detection performance; as the correlation increases in the training set, OOD detection performance severely worsens [74] [72].
The Scientist's Toolkit: Essential Research Reagents & Platforms
Table 2: Key Research Reagents and Platforms for OOD Molecular Research
| Tool Name | Type | Primary Function in OOD Research | Access |
|---|---|---|---|
| BOOM Benchmark [9] | Benchmark Suite | Standardized evaluation of OOD generalization for molecular property prediction models. | Open-source (GitHub) |
| ODP-Bench [75] | Benchmark Suite | Provides 1,444 trained models and 29 datasets for benchmarking OOD performance prediction algorithms. | Open-source (GitHub) |
| Baishenglai (BSL) [76] | Integrated Platform | An end-to-end drug discovery platform emphasizing OOD generalization across 7 core tasks (e.g., DTI, generation). | Publicly accessible (web) |
| EviDTI [77] | Prediction Framework | A Drug-Target Interaction (DTI) prediction model using Evidential Deep Learning to provide reliable uncertainty estimates for OOD data. | Open-source (GitHub) |
| QM9, 10K Datasets [9] | Data | Curated molecular datasets with quantum chemical properties, used as base data for constructing OOD splits. | Public |
| Nuisance-Randomized Distillation (NURD) [72] | Algorithm | Trains classifiers to be robust to spurious correlations by learning from a distribution where the nuisance-label relationship is broken. | Methodological |
The correlation between in-distribution performance and out-of-distribution success is weak and unreliable. Current benchmarks demonstrate that even state-of-the-art models suffer from a significant performance gap when facing OOD data. Success depends critically on the nature of the distribution shift and the model's ability to avoid learning spurious correlations. For drug discovery researchers, prioritizing models and platforms that incorporate robust OOD evaluation, uncertainty quantification, and explicit strategies for mitigating spurious features is essential for translating computational predictions into real-world therapeutic breakthroughs. The future of reliable molecular property prediction lies not merely in optimizing ID accuracy, but in building models with explicitly designed OOD robustness.
Conclusion
Achieving robust out-of-distribution generalization remains a paramount, unsolved challenge in molecular property prediction, as current models, including advanced GNNs and transformers, exhibit significantly higher error rates on OOD data. However, promising pathways forward have emerged. Methodological innovations in transduction, meta-learning, and sophisticated uncertainty quantification offer tangible improvements in extrapolation accuracy and reliability. The development of rigorous, standardized benchmarks like BOOM is crucial for meaningful progress, providing the tools for unbiased comparative analysis. For biomedical and clinical research, the implications are profound. Prioritizing OOD robustness in model development and selection is not merely an academic exercise but a necessary step to de-risk the drug discovery pipeline, ensuring that computational predictions are reliable when they matter most—for novel, groundbreaking compounds that truly expand the boundaries of known chemistry. Future efforts must focus on creating more chemically-aware architectures, developing better methods for leveraging vast unlabeled datasets, and establishing universal benchmarking standards to build foundation models that generalize reliably across the vast expanse of chemical space.
References
- 1. Known Unknowns: Out-of-Distribution Property Prediction in Materials... [https://arxiv.org/html/2502.05970v1]
- 2. Out-of-Distribution Property Prediction in Materials and ... [https://www.nature.com/articles/s41524-025-01808-x]
- 3. Optimizing OOD Detection in Molecular Graphs: A Novel ... [https://arxiv.org/html/2404.15625v1]
- 4. Consistent semantic representation learning for out-of-distribution... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11982020/]
- 5. Out of Distribution Detection: Knowing When AI Doesn't Know [https://www.sei.cmu.edu/blog/out-of-distribution-detection-knowing-when-ai-doesnt-know/]
- 6. Reducing overconfident errors in molecular property ... [https://www.sciencedirect.com/science/article/pii/S2666389924001065]
- 7. Towards a more inductive world for drug repurposing ... [https://www.nature.com/articles/s42256-025-00987-y]
- 8. Robustness in deep learning models for medical diagnostics [https://link.springer.com/article/10.1007/s10462-024-11005-9]
- 9. BOOM: Benchmarking Out-Of-distribution Molecular ... [https://arxiv.org/html/2505.01912v1]
- 10. Structure-based out-of-distribution (OOD) materials ... [https://www.nature.com/articles/s41524-024-01316-4]
- 11. BOOM: Benchmarking Out-Of-distribution Molecular ... [https://arxiv.org/abs/2505.01912]
- 12. BOOM: Benchmarking Out-Of-distribution Molecular ... [https://neurips.cc/virtual/2025/poster/121661]
- 13. A BENCHMARK AND EVALUATION FOR REAL-WORLD ... [https://arxiv.org/html/2501.18463v1]
- 14. A Benchmark and Evaluation for Real-World Out-of- ... [https://arxiv.org/abs/2501.18463]
- 15. OpenMIBOOD: Open Medical Imaging Benchmarks for Out-Of ... [https://cvpr.thecvf.com/virtual/2025/poster/33159]
- 16. Evaluating Machine Learning Models for Molecular ... [https://chemrxiv.org/engage/chemrxiv/article-details/67c8a90bfa469535b9148866]
- 17. Molecular property prediction in the ultra‐low data regime [https://www.nature.com/articles/s42004-025-01592-1]
- 18. Handling Out-of-Distribution Data: A Survey [https://arxiv.org/html/2507.21160v1]
- 19. Characterizing Out-of-Distribution Error via Optimal Transport [https://arxiv.org/abs/2305.15640]
- 20. Inductive vs. Transductive Learning [https://medium.com/data-science/inductive-vs-transductive-learning-e608e786f7d]
- 21. A Transductive Approach in Offline Reinforcement Learning [https://arxiv.org/html/2404.04682v1]
- 22. Positional embeddings and zero-shot learning using BERT for ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-00959-9]
- 23. Robust Molecular Property Prediction via Densifying ... [https://arxiv.org/html/2506.11877v1]
- 24. Probing out-of-distribution generalization in machine ... [https://www.nature.com/articles/s43246-024-00731-w]
- 25. Context-informed few-shot molecular property prediction ... [https://www.sciencedirect.com/science/article/pii/S2001037025003381]
- 26. A new method of semi-supervised learning classification ... [https://www.nature.com/articles/s41598-025-02324-0]
- 27. A Survey on Evaluation of Out-of-Distribution Generalization [https://arxiv.org/html/2403.01874v1]
- 28. A critical review of machine learning interatomic potentials ... [https://www.oaepublish.com/articles/jmi.2025.17]
- 29. E(3) equivariant graph neural networks for robust and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10499216/]
- 30. Advancing molecular property prediction for environmental ... [https://www.sciencedirect.com/science/article/pii/S0169743925002667?dgcid=rss_sd_all]
- 31. Accelerating materials property prediction via a hybrid ... [https://www.nature.com/articles/s41524-024-01472-7]
- 32. Graph & Geometric ML in 2024: Where We Are and What's ... [https://towardsdatascience.com/graph-geometric-ml-in-2024-where-we-are-and-whats-next-part-i-theory-architectures-3af5d38376e1/]
- 33. A quantum-transformer hybrid architecture for polymer ... [https://www.sciencedirect.com/science/article/pii/S0927025625002939?dgcid=rss_sd_all]
- 34. Context-informed few-shot molecular property prediction via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12510055/]
- 35. Real-World Molecular Out-Of-Distribution: Specification and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10865358/]
- 36. ChemBERTa embeddings and ensemble learning for ... [https://www.sciencedirect.com/science/article/abs/pii/S0098135425000699]
- 37. Adaptive Individual Uncertainty under Out-Of-Distribution ... [https://arxiv.org/html/2510.15233v1]
- 38. Robust Molecular Property Prediction via Densifying ... [https://arxiv.org/abs/2506.11877]
- 39. Understanding covariate in model performance | F1000Research shift [https://f1000research.com/articles/5-597]
- 40. : Dataset Covariate in Machine Learning Shift Shift [https://www.analyticsvidhya.com/blog/2017/07/covariate-shift-the-hidden-problem-of-real-world-data-science/]
- 41. Evaluating Machine Learning Models for Molecular ... [https://chemrxiv.org/engage/chemrxiv/article-details/67c0bc4c6dde43c908c2a714]
- 42. Scaffold-Aware Generative Augmentation and Reranking ... [https://arxiv.org/html/2510.16306v1]
- 43. Reducing overconfident errors in molecular property ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11240180/]
- 44. Evidential Deep Learning for Guided Molecular Property ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8393200/]
- 45. Integrating Explainability and Uncertainty Estimation in ... [https://arxiv.org/html/2509.18132v1]
- 46. Uncertainty Estimation by Flexible Evidential Deep Learning [https://arxiv.org/html/2510.18322v1]
- 47. [2505.08709] Contrastive Normalizing for Flows -Aware... Uncertainty [https://arxiv.org/abs/2505.08709]
- 48. Multi-task methods for molecular property prediction [https://www.sciencedirect.com/science/article/pii/S0098135425002571]
- 49. On the Connection between Pre-training Data Diversity ... [https://arxiv.org/abs/2307.12532]
- 50. Language Models Improve When Pretraining Data ... [https://arxiv.org/abs/2507.12466]
- 51. Ask Your Distribution Shift if Pre-Training is Right for You [https://gradientscience.org/pretraining-robustness/]
- 52. Exploring Data Splitting Strategies for Offline Evaluation of ... [https://arxiv.org/abs/2507.16289]
- 53. Mastering Data Splitting in Data Science: Techniques for ... [https://medium.com/@adnan.mazraeh1993/mastering-data-splitting-in-data-science-techniques-for-robust-model-evaluation-and-performance-48df94a197ca]
- 54. Molecular Connectivity Index-Based Data Augmentation for ... [https://pubmed.ncbi.nlm.nih.gov/40811367/]
- 55. LLM Training Methodologies in 2025: Pretraining, Fine ... [https://klizos.com/llm-training-methodologies-in-2025/]
- 56. Hyperparameter Tuning: Complete 2025 Guide To ML ... [https://www.futureskillguides.com/hyperparameter-tuning-guide-2025/]
- 57. Methodology for hyperparameter tuning of deep neural ... [https://www.sciencedirect.com/science/article/abs/pii/S0098135424003466]
- 58. Multi-objective Hyperparameter Optimization in the Age of ... [https://arxiv.org/html/2511.08371v1]
- 59. Dynamic robustness evaluation for automated model ... [https://www.sciencedirect.com/science/article/pii/S0950584924002088]
- 60. M3OOD: Automatic Selection of Multimodal OOD Detectors [https://arxiv.org/abs/2508.11936]
- 61. Kolmogorov–Arnold graph neural networks for molecular ... [https://www.nature.com/articles/s42256-025-01087-7]
- 62. Robust Molecular Property Prediction via Densifying ... [https://ui.adsabs.harvard.edu/abs/2025arXiv250611877K/abstract]
- 63. Physical encoding improves OOD performance in deep ... [https://www.sciencedirect.com/science/article/abs/pii/S0927025624008243]
- 64. Evaluating Machine Learning Models for Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12529777/]
- 65. Advancing ADMET prediction through multiscale fragment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12478026/]
- 66. Evaluating Machine Learning Models for Molecular ... [https://pubmed.ncbi.nlm.nih.gov/40947919/]
- 67. Benchmarking ML in ADMET predictions: the practical impact ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01041-0]
- 68. ADMET evaluation in drug discovery: 21. Application and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11724520/]
- 69. Graph neural networks driven acceleration in drug discovery [https://www.sciencedirect.com/science/article/pii/S2211383525006884]
- 70. Revolutionizing pharmacology: AI-powered approaches in ... [https://www.sciencedirect.com/science/article/pii/S259009862500020X]
- 71. Transformer models in biomedicine - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11287876/]
- 72. Robustness to Spurious Correlations Improves Semantic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10923583/]
- 73. Out-of-distribution detection with non-semantic exploration [https://www.sciencedirect.com/science/article/pii/S0020025525001215]
- 74. On the Impact of Spurious Correlation for Out-of-distribution ... [https://arxiv.org/abs/2109.05642]
- 75. Benchmarking Out-of-Distribution Performance Prediction [https://arxiv.org/html/2510.27263v1]
- 76. Empowering Drug Discovery through Intelligent, Multitask ... [https://arxiv.org/html/2508.01195v1]
- 77. Evidential deep learning-based drug-target interaction ... [https://www.nature.com/articles/s41467-025-62235-6]