Semi-Empirical Methods for IR Spectra Simulation: A Practical Guide for Biomedical Researchers
This comprehensive review explores semi-empirical quantum mechanical methods for simulating infrared (IR) spectra, addressing critical needs in drug discovery and materials science.
Semi-Empirical Methods for IR Spectra Simulation: A Practical Guide for Biomedical Researchers
Abstract
This comprehensive review explores semi-empirical quantum mechanical methods for simulating infrared (IR) spectra, addressing critical needs in drug discovery and materials science. Covering foundational theories to cutting-edge machine learning integrations, we examine popular methodologies (AM1, PM6, PM7, GFN-xTB) and their practical applications in analyzing functionalized nanomaterials, metal complexes, and pharmaceutical compounds. The article provides troubleshooting guidance for accuracy limitations and comparative validation against experimental data and higher-level theoretical methods. Special emphasis is placed on interpreting 2D material functionalization, tautomer identification, and protonation state analysis—crucial challenges in biomedical research where traditional IR assumptions often fail.
Understanding Semi-Empirical IR Simulation: Core Principles and Method Evolution
Semi-empirical quantum mechanical methods occupy a crucial niche in computational chemistry, providing a balance between computationally intensive ab initio methods and simpler molecular mechanics approaches. These methods achieve their computational efficiency by using approximations to bypass costly calculations and employing empirically determined parameters optimized against reference data. Among the various approximations, the Neglect of Diatomic Differential Overlap (NDDO) family has emerged as one of the most robust and widely used frameworks. The NDDO approach forms the foundation for popular semi-empirical methods including MNDO, AM1, PM3, PM6, and the more recent PM7, each representing successive refinements in both approximation and parameterization strategies.
The fundamental approximation in NDDO methods involves neglecting differential overlap terms between atomic orbitals located on different atoms, while retaining those on the same atom. This significant simplification reduces the computational complexity of the two-electron integrals that constitute the most challenging aspect of quantum chemical calculations. Modern implementations have further refined these approximations to address specific limitations, particularly for modeling non-covalent interactions and solid-state systems where earlier implementations exhibited significant errors. The continued evolution of NDDO methods demonstrates their enduring value in modeling large and complex molecular systems where higher-level calculations remain computationally prohibitive.
Theoretical Foundations of the NDDO Approximation
Core Mathematical Formalism
The NDDO approximation operates within the Löwdin orthogonalized basis set, where the overlap matrix between basis functions is replaced by the identity matrix. This fundamental simplification dramatically streamlines the solution of the Hartree-Fock equations. Within this framework, the Fock matrix elements are constructed from one-electron (( \mathbf{H} )) and two-electron (( \mathbf{G} )) components, with the NDDO approximation specifically affecting the treatment of two-electron integrals.
The key NDDO approximation involves neglecting all two-center two-electron integrals unless the orbitals are located on the same atom. This means that integrals of the form ( (\mu\nu|\lambda\sigma) ) are retained only when orbitals ( \mu ) and ( \nu ) are on the same atomic center AND orbitals ( \lambda ) and ( \sigma ) are on the same atomic center (which may be different from the first). All other integral types are set to zero. This approximation significantly reduces the number of two-electron integrals that must be computed, providing substantial computational savings while retaining some degree of quantum mechanical rigor.
The general Coulomb and exchange matrices within this framework can be defined as follows. The Coulomb matrix ( \mathbf{J}({\varvec{\Delta}}) ) for an arbitrary density matrix ( {\varvec{\Delta}} ) is given by:
[ {J}{\mu\nu}({\varvec{\Delta}}) = \begin{cases} \sum{\lambda \in A} {\Delta}{\lambda\lambda} (\mu\nu|\lambda\lambda) + \sum{\lambda,\sigma \in B \ne A} {\Delta}{\lambda\sigma} (\mu\nu|\lambda\sigma), & \mu = \nu, \mu,\nu \in A \ 2{\Delta}{\mu\nu} (\mu\nu|\mu\nu) + \sum{\lambda,\sigma \in B \ne A} {\Delta}{\lambda\sigma} (\mu\nu|\lambda\sigma), & \mu,\nu \in A \ 0, & \mu \in A, \nu \in B \ne A \end{cases} ]
Similarly, the exchange matrix ( \mathbf{K}({\varvec{\Lambda}}) ) for an arbitrary matrix ( {\varvec{\Lambda}} ) is defined as:
[ {K}{\mu\nu}({\varvec{\Lambda}}) = \begin{cases} \sum{\lambda \in A} {\Lambda}{\lambda\lambda} (\mu\lambda|\nu\lambda), & \mu = \nu, \mu,\nu \in A \ {\Lambda}{\mu\nu} \left[ (\mu\nu|\mu\nu) + (\mu\mu|\nu\nu) \right], & \mu,\nu \in A \ 0, & \mu \in A, \nu \in B \ne A \end{cases} ]
These definitions illustrate how the NDDO approximation maintains the integrity of one-center terms while simplifying two-center interactions [1].
Evolution of NDDO Approximations
Modern NDDO implementations have addressed several limitations observed in earlier versions. In particular, PM7 introduced critical modifications to improve the description of noncovalent interactions and rectified two minor errors in the original NDDO formalism. A key enhancement involves constraints on the core-core interaction terms to ensure physically realistic behavior at large interatomic separations.
In conventional NDDO methods, the rate at which nuclear-nuclear interactions converged to exact values at large distances differed depending on the specific atoms involved. While chemically insignificant for molecular systems, these small differences produced infinite errors when applied to crystalline solids. To address this, a modified potential was implemented where the two-electron two-center integral ( \gamma_{AB} ) (normally abbreviated from ( \langle ss|ss \rangle )) transitions smoothly to the exact point charge expression at 7.0 Å, well beyond covalent bonding distances:
[ \gamma{AB} = \frac{1}{R{AB} + \frac{1}{2}(GA + GB)} ]
where ( G_A ) is the two-electron one-center integral for atom A. This modification ensures that electron-electron repulsion, electron-nuclear attraction, and nuclear-nuclear repulsion terms are exactly balanced beyond 7 Å, eliminating spurious long-range attractions or repulsions between neutral atoms [2].
Additionally, constants were added to the multipole integrals of type ( \langle pp|pp \rangle ) and related terms to ensure their average values match the ( \langle ss|ss \rangle ) integral. This correction addresses spurious contributions to solid-state energies arising from hybrid orbitals or lone pairs, further improving the method's applicability to extended systems [2].
Parameterization Strategies for NDDO Methods
Parameter Optimization Framework
The accuracy of NDDO-based semi-empirical methods is critically dependent on the parameterization process, which optimizes atomic parameters to reproduce reference data. The standard approach minimizes an error function ( \mathcal{S} ) that measures the difference between calculated and reference molecular properties:
[ \mathcal{S} = \sum{\alpha} \mathcal{C}{\alpha}^2 \left( \xi{\alpha}^{\text{ref}} - \xi{\alpha} \right)^2 ]
where ( \xi{\alpha} ) represents calculated properties, ( \xi{\alpha}^{\text{ref}} ) their reference values, and ( \mathcal{C}_{\alpha} ) weighting factors that prioritize different property types.
The optimization process requires calculating first and second derivatives of this error function with respect to the parameters. The second derivative (Hessian) matrix provides critical information about the nature of stationary points during optimization:
[ \frac{\partial^2 \mathcal{S}}{\partial {}^{ZA}pi \partial {}^{ZB}pj} = 2\sum{\alpha} \mathcal{C}{\alpha}^2 \left[ \left( \xi{\alpha} - \xi{\alpha}^{\text{ref}} \right) \frac{\partial^2 \xi{\alpha}}{\partial {}^{ZA}pi \partial {}^{ZB}pj} + \frac{\partial \xi{\alpha}}{\partial {}^{ZA}pi} \frac{\partial \xi{\alpha}}{\partial {}^{ZB}p_j} \right] ]
where ( {}^{ZA}pi ) represents parameter ( i ) for element ( Z_A ). Recent work has revealed that some parameterization schemes, including that used for PM7, neglected the second-derivative term of the reference functions, potentially affecting optimization quality [1].
Analytical Derivative Methods
Efficient parameter optimization benefits greatly from analytical methods for evaluating parameter derivatives of molecular properties. This requires solving coupled-perturbed Hartree-Fock (CPHF) equations, which become simplified in the Löwdin basis where the overlap matrix is identity. The first-order CPHF equations are well-established in NDDO methods, but recent advances have extended these to second order, enabling more robust parameter optimization.
The second-order CPHF equations account for how changes in parameters affect the electronic structure through the density matrix. For Unrestricted Hartree-Fock (UHF) formalisms, these equations must be solved separately for alpha- and beta-spin density matrices, with the total density matrix ( \mathbf{P} = {}^{\alpha}\mathbf{P} + {}^{\beta}\mathbf{P} ) representing the sum of both components. Implementation of these analytical derivatives addresses numerical instability issues associated with finite-difference approaches and ensures reproducibility in parameter optimization [1].
Reference Data Selection
The choice of reference data significantly impacts parameterization outcomes. As noted in the development of PM7, "the origins of the errors in NDDO methods have been examined, and were found to be attributable to inadequate and inaccurate reference data" [2]. Modern parameterization increasingly emphasizes diverse molecular classes, including both organic compounds and coordination complexes, to enhance method transferability.
PM7 incorporated a new type of reference data intended to better define the structure of parameter space, alongside traditional experimental and high-level ab initio reference data. This approach yielded significant improvements: for simple gas-phase organic systems, the average unsigned error (AUE) in bond lengths decreased by approximately 5% and the AUE in heats of formation (( \Delta Hf )) decreased by about 10% compared to PM6. For organic solids, the improvement was even more dramatic, with AUE in ( \Delta Hf ) dropping by 60% and geometric errors reduced by 33.3% [2].
Table 1: Performance Comparison of NDDO Methods for Different System Types
| System Type | Property | PM6 AUE | PM7 AUE | Improvement |
|---|---|---|---|---|
| Organic Molecules | Bond Lengths | Baseline | ~5% reduction | ~5% |
| Organic Molecules | ΔHf | Baseline | ~10% reduction | ~10% |
| Organic Solids | ΔHf | Baseline | ~60% reduction | ~60% |
| Organic Solids | Geometries | Baseline | ~33.3% reduction | ~33.3% |
| Organic Reactions | Barrier Heights | 12.6 kcal/mol | 10.8 kcal/mol | 14.3% |
| Organic Reactions (PM7-TS) | Barrier Heights | 12.6 kcal/mol | 3.8 kcal/mol | 69.8% |
Experimental Protocols for IR Spectra Simulation
Geometry Optimization Protocol
Accurate simulation of infrared spectra requires careful geometry optimization as a foundational step. The following protocol outlines the standard procedure for preparing molecular structures for vibrational frequency calculations:
Initial Structure Construction: Build the molecular structure using chemical drawing software or coordinate generators. For regular polyaromatic hydrocarbons, initial geometries may assume standard bond lengths (e.g., C-C distance of 1.397 Å, C-H distance of 1.084 Å, and bond angles of 120°) [3].
Preliminary Optimization: Perform an initial geometry optimization using molecular mechanics methods to remove severe steric clashes and obtain a reasonable starting structure for quantum chemical calculations.
Semi-Empirical Optimization: Conduct full geometry optimization using an NDDO method (AM1, PM3, PM6, or PM7) with the following typical parameters:
- Basis set: Unrestricted Hartree-Fock for open-shell systems, Restricted Hartree-Fock for closed-shell systems
- Optimization algorithm: Polak-Ribiere conjugate gradient or similar
- Convergence limit: 0.001 kcal/mol or 0.05 kcal/mol/Ångstrom for the energy gradient [3] [4]
- Self-consistent field (SCF) convergence: Typically 10⁻⁵ to 10⁻⁶ au
Convergence Verification: Confirm that the optimization has reached a true minimum by verifying that the gradient norm is below the threshold and examining the resulting molecular structure for chemical reasonableness.
Vibrational Frequency Calculation
Once an optimized geometry is obtained, vibrational frequencies and infrared intensities can be calculated:
Force Constant Calculation: Compute the second derivative matrix (Hessian) of the energy with respect to nuclear coordinates at the optimized geometry. This is typically performed analytically where available or via numerical differentiation of analytical gradients.
Frequency Determination: Diagonalize the mass-weighted Hessian matrix to obtain vibrational frequencies. The eigenvalues correspond to squared vibrational frequencies, which are converted to wavenumbers (cm⁻¹).
IR Intensity Calculation: Compute the infrared intensities as the derivative of the dipole moment with respect to normal coordinates. This provides the relative strength of absorption for each vibrational mode.
Frequency Scaling: Apply scaling factors to account for systematic errors inherent in the NDDO methods. These factors are typically determined empirically by comparing calculated frequencies with experimental reference data for standard compounds.
Spectral Assignment: Analyze the vibrational normal modes through visualization to assign spectral features to specific molecular motions. Modern software packages provide animation capabilities that display the nuclear motions associated with each vibrational mode, greatly facilitating assignment [3] [4].
Table 2: Computational Methods for IR Spectral Simulation
| Method | Theoretical Basis | Speed | Accuracy | Best Applications |
|---|---|---|---|---|
| AM1 | NDDO-based semi-empirical | Very Fast | Moderate | Organic molecules, functional group identification |
| PM3 | NDDO-based semi-empirical | Very Fast | Moderate | Organic molecules, metal complexes |
| PM6 | NDDO-based semi-empirical | Very Fast | Good | Organic molecules, solids |
| PM7 | NDDO-based semi-empirical | Very Fast | Good | Non-covalent interactions, solids |
| DFT | First principles | Moderate | High | Accurate spectral prediction |
| AIQM | ML-corrected semi-empirical | Fast | Very Good | Balanced accuracy and speed |
Electronic Spectrum Calculation (ZINDO Protocol)
For complete spectroscopic characterization, electronic excitation spectra can be calculated using Zerner's Intermediate Neglect of Differential Overlap (ZINDO) method:
Ground State Calculation: Perform a single-point calculation on the AM1-optimized geometry to obtain molecular orbital coefficients and eigenvalues.
Configuration Interaction Setup: Define the active space for configuration interaction (CI) calculations, typically including single excitations from occupied to virtual orbitals.
Transition Energy Calculation: Compute singlet-singlet transition energies using the formula: [ \Delta E{ia} = \varepsilona - \varepsiloni - J{ia} + 2K{ia} ] where ( \varepsilona ) and ( \varepsiloni ) are orbital energies, ( J{ia} ) is the molecular Coulomb integral ( (ii|aa) ), and ( K_{ia} ) is the molecular exchange integral ( (ia|ai) ) [3].
Spectrum Generation: Construct the electronic spectrum by calculating oscillator strengths for each transition and applying appropriate broadening functions to simulate band shapes.
Workflow for NDDO-Based IR Spectrum Simulation
Applications in Molecular Spectroscopy
IR Spectroscopy of Organic Molecules and 2D Materials
NDDO-based semi-empirical methods have proven particularly valuable for simulating infrared spectra of complex molecular systems, including two-dimensional materials like graphene quantum dots (GQDs). These materials present unique challenges for spectroscopic analysis because traditional IR interpretation assumes three-dimensional materials, making it difficult to identify functional groups located at edges or center surfaces of 2D structures.
Studies on pyrene-like molecules as models for GQDs demonstrate how semi-empirical methods can elucidate structure-spectra relationships in these systems. The AM1 method has shown success in predicting infrared intensities and spectral patterns for GQDs, particularly in the aromatic vibration region (1400-2000 cm⁻¹) corresponding to C=C stretching modes, and in the 3000-3500 cm⁻¹ region for C-H and O-H stretching vibrations. This capability provides crucial insights for identifying edge and center surface functionalization in 2D carbon materials [3].
Functionalization significantly affects the vibrational spectra of 2D materials. For instance, introducing oxygen-containing groups (hydroxyl, epoxy) or nitrogen-containing groups (amine, pyridine) creates distinctive spectral signatures that can be identified through semi-empirical simulations. These computational approaches complement experimental techniques like FTIR and XPS, enabling more precise correlation between spectral features and specific functionalization patterns [3].
Metal Complexes and Pharmaceutical Applications
The application of NDDO methods extends to metal complexes of pharmaceutical compounds, where IR spectroscopy assists in characterizing coordination modes and ligand binding. Studies on cobalt and cadmium complexes of sulfanilamide demonstrate how PM3 simulations can validate structures proposed based on experimental IR spectra and X-ray powder diffraction patterns.
Coordination effects on vibrational modes of functional groups (e.g., anilino and sulfonamido groups in sulfanilamide) can be accurately reproduced by semi-empirical methods, providing atomic-level insights into metal-ligand interactions. The visualization of vibrational normal modes through animation capabilities in modern software packages greatly facilitates interpretation of these complex spectra [4].
For drug development applications, semi-empirical IR simulations offer a rapid screening tool for predicting spectroscopic properties of metal-drug complexes, which often exhibit enhanced biological activity compared to the parent drugs. The ability to simulate how coordination affects vibrational frequencies helps identify binding modes and assess complex stability [4].
Research Toolkit
Table 3: Essential Computational Tools for NDDO-Based Spectroscopy
| Tool/Resource | Type | Key Functionality | Application in IR Spectroscopy |
|---|---|---|---|
| MOPAC | Software Package | NDDO method implementation (MNDO, AM1, PM3, PM6, PM7) | Geometry optimization, frequency calculation, thermodynamic properties |
| Winmostar | Software Package | AM1 and ZINDO methods with graphical interface | Vibrational frequency calculation, electronic spectrum simulation |
| HyperChem | Software Package | Semi-empirical methods with visualization | Geometry optimization, IR simulation, vibrational mode animation |
| MLatom | Software Package | AIQM and machine learning methods | Enhanced accuracy IR spectra with semi-empirical speed |
| ZINDO Method | Computational Method | Spectroscopic parameterization for electronic transitions | UV-Vis spectrum simulation, complementary to IR analysis |
| PM7 Parameters | Parameter Set | Optimized for non-covalent interactions and solids | Improved accuracy for complex molecular systems |
The NDDO approximation continues to provide a robust foundation for semi-empirical quantum chemical methods nearly seven decades after its initial development. Recent advances in parameterization strategies, particularly improved treatment of long-range interactions and more sophisticated optimization of empirical parameters, have significantly expanded the applicability of NDDO-based methods to diverse chemical systems including organic molecules, coordination compounds, and extended materials.
For IR spectroscopic applications, methods like AM1, PM3, PM6, and PM7 offer a compelling balance between computational efficiency and predictive accuracy. The protocols outlined in this document provide researchers with standardized approaches for leveraging these methods in spectroscopic analysis of molecular structure and functional group identification. As machine learning approaches begin to complement traditional semi-empirical methods, exemplified by the AIQM family of models, the future promises even more accurate and efficient simulation of molecular spectra while retaining the interpretability that makes quantum chemical methods scientifically valuable.
NDDO Method Evolution and Applications
Semi-empirical quantum chemistry methods occupy a crucial niche in computational chemistry, providing an intermediate level of theory between computationally intensive ab initio methods and simpler molecular mechanics approaches. By employing fundamental quantum mechanical principles augmented with empirically derived parameters, these methods achieve a balance of computational efficiency and accuracy that enables the study of large molecular systems. The evolution of these methods—from the early Modified Neglect of Diatomic Overlap (MNDO) through Austin Model 1 (AM1) to the parametric methods PM6 and PM7—represents a continuous endeavor to expand their applicability and reliability while maintaining their computational advantages. Within the specific context of infrared (IR) spectra simulation, these methods have proven particularly valuable for researchers studying complex molecular systems, including those in drug development, where understanding vibrational signatures is essential for characterizing molecular structure and interactions.
Historical Development of Semi-Empirical Methods
The development of semi-empirical methods follows a logical progression, with each generation building upon and addressing limitations of its predecessors. The foundational approximation for most modern semi-empirical methods is the Neglect of Diatomic Differential Overlap (NDDO), which significantly reduces computational complexity by approximating certain electron integrals [5].
The historical trajectory of these methods began with MNDO, which was later refined to create AM1. The Austin Model 1 (AM1) introduced Gaussian functions to the core-core repulsion terms in the Hamiltonian, leading to improved description of short-range interactions compared to its predecessor [6]. AM1 has demonstrated particular utility in IR spectra simulation, successfully predicting infrared intensities and spectral patterns for organic materials, with results showing agreement with experimental data [3].
The next significant advancement came with PM6 (Parametric Method 6), which incorporated diatomic parameters rather than the element-specific parameters used in AM1, and also included parameters for d-orbitals [6]. This expansion provided improved treatment of a wider range of chemical elements and bonding situations.
The most recent method in this lineage, PM7 (Parametric Method 7), represents a substantial refinement through the inclusion of specific corrections for noncovalent interactions, particularly dispersion forces and hydrogen bonding [6] [7]. PM7 also rectified two minor errors in the NDDO formalism and introduced a modified treatment of core-core interactions that eliminated infinite errors when modeling crystalline solids [7] [8]. This correction made PM7 uniquely capable among semi-empirical methods for reliably predicting crystal structures and heats of formation of solids [8].
Table: Historical Evolution of Key Semi-Empirical Methods
| Method | Key Innovations | Strengths | Known Limitations |
|---|---|---|---|
| MNDO | Original NDDO implementation; parameterized for heats of formation | Foundation for later methods | Limited accuracy for noncovalent interactions |
| AM1 | Added Gaussian functions to core-core repulsion | Improved short-range interactions vs MNDO; useful for IR spectra simulation [3] | Remaining deficiencies in hydrogen bonding |
| PM6 | Used diatomic parameters; included d-orbital parameters | Better accuracy for geometries and ΔHf vs predecessors [7] | Errors in certain atomic pair repulsions [7] |
| PM7 | Added dispersion and hydrogen bond corrections; fixed solid-state electrostatic errors | Vastly improved for noncovalent interactions and solids [7] [8] | Parameterized for limited elements (like PM6) |
Performance Benchmarking and Accuracy Assessment
Rigorous benchmarking against experimental data and higher-level theoretical calculations provides crucial insights into the performance characteristics of semi-empirical methods. Extensive benchmark studies have evaluated these methods for general ground-state properties including energies (atomization energies, reaction energies, heats of formation, barrier heights) and molecular geometries [9].
For organic compounds, PM7 shows measurable improvements over PM6. The average unsigned error (AUE) in bond lengths decreased by approximately 5%, while the AUE in heats of formation (ΔHf) decreased by about 10% [7]. The improvements are even more pronounced for organic solids, where PM7 reduced errors in ΔHf by 60% and geometric errors by 33.3% compared to PM6 [7]. For reaction barrier heights, a two-step process called PM7-TS significantly improved accuracy, reducing the AUE from 12.6 kcal/mol in PM6 and 10.8 kcal/mol in PM7 to 3.8 kcal/mol [7].
In the specific context of soot formation simulations, which involve polycyclic aromatic hydrocarbons relevant to IR spectroscopy applications, GFN2-xTB demonstrated the best performance among semi-empirical methods, followed by DFTB3 and DFTB2 [6]. The performance of PM6 and PM7 was found to be similar, with no clear improvement observed in PM7 over PM6 for these systems [6].
Table: Comparative Performance Metrics for Semi-Empirical Methods
| Property | AM1 | PM6 | PM7 | GFN2-xTB |
|---|---|---|---|---|
| Bond Length AUE | - | Baseline | ~5% improvement [7] | - |
| ΔHf AUE (organic) | - | Baseline | ~10% improvement [7] | - |
| ΔHf AUE (solids) | - | Baseline | ~60% improvement [7] | - |
| Barrier Height AUE | - | 12.6 kcal/mol | 10.8 kcal/mol (3.8 with PM7-TS) [7] | - |
| Noncovalent Interactions | Limited | Moderate | Good with explicit corrections [7] | Good with D4 dispersion [6] |
| IR Spectrum Prediction | Successful for organic materials [3] | - | - | - |
Application to IR Spectra Simulation: Protocols and Procedures
IR Spectral Simulation of Pyrene-like Molecules Using AM1
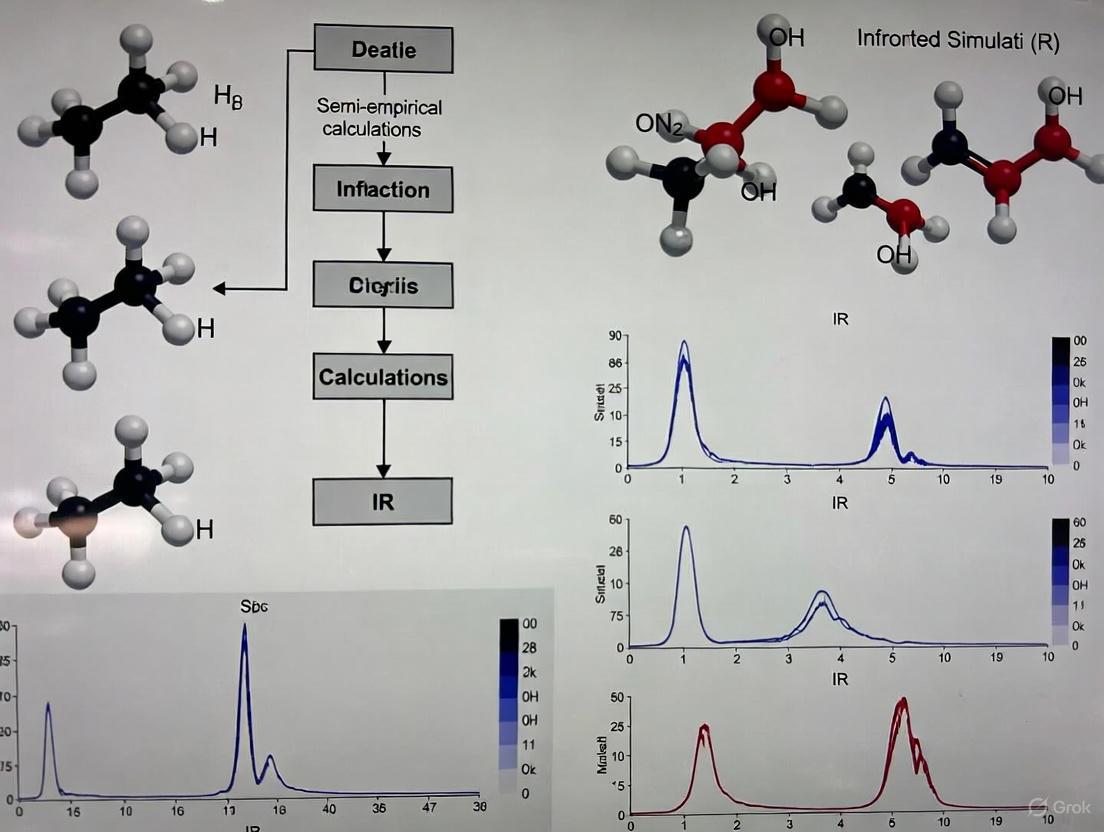
The application of semi-empirical methods to IR spectra simulation provides a computationally efficient approach for characterizing complex molecular systems. A representative protocol for simulating IR spectra of pyrene-like molecules as models for graphene quantum dots (GQDs) using the AM1 method involves the following steps [3]:
Initial Structure Preparation: Construct the initial molecular geometry using standard bond lengths and angles. For polycyclic aromatic hydrocarbons like pyrene, this typically involves regular networks with C-C distances of 1.397 Å, C-H distances of 1.084 Å, and bond angles of 120°.
Geometry Optimization:
- Perform preliminary optimization using molecular mechanics methods.
- Refine the geometry using the AM1 semi-empirical method with a convergence limit based on orientation observations, typically reaching a gradient limit of the energy change of 0.05 kcal/mol/Ångstrom.
- The geometry optimization employs a second-order Taylor energy expansion around the current point.
Vibrational Frequency Calculation:
- After obtaining a stable optimized structure, perform a force calculation to obtain vibrational modes.
- Compare results against experimental data for validation.
- If the match between calculated and experimental frequencies exceeds 10% error, consider the semi-empirical technique unsuitable for that specific vibrational mode.
Spectral Analysis:
- Focus analysis on the aromatics vibrations region (1400-2000 cm⁻¹) for C=C stretching modes.
- Examine the 3000-3500 cm⁻¹ region for C-H and O-H stretching vibrations in functionalized systems.
- For functionalized GQDs, add functional groups (e.g., hydroxyl, methyl) to both edge and center positions of the molecular surface to assess positional effects on spectral features.
This protocol has demonstrated particular utility for analyzing 2D carbon materials like graphene quantum dots, where conventional IR spectroscopy assuming 3D materials struggles to distinguish functional groups located at edges versus the center of the 2D surface [3].
Diagram 1: Workflow for IR spectra simulation using the AM1 semi-empirical method
Conformational Analysis Using DFTB3 for IR Spectrum Assignment
For the assignment of experimental IR spectra to specific molecular conformers, the DFTB3 semi-empirical method has proven valuable in the initial stages of conformational analysis:
Potential Energy Surface Scanning:
- Employ DFTB3 to efficiently scan the potential energy surface of the target molecule.
- Identify low-energy conformers as candidates for further analysis.
- This approach represents a good compromise between accuracy and computational cost for this initial screening phase [10].
Conformer Pre-optimization:
- Utilize density functional theory with generalized gradient approximation (GGA) functionals and a small basis set including polarization functions.
- This level of theory provides sufficient accuracy while maintaining computational efficiency for the pre-optimization step [10].
Energy-Based Conformer Selection:
- Apply hybrid functionals for more accurate energy evaluations of pre-optimized conformers.
- Use an energy window of at least 15 kJ/mol when selecting conformers for spectral comparison to ensure inclusion of potentially relevant structures [10].
Spectral Comparison and Assignment:
- Compare calculated IR spectra of candidate conformers with experimental data.
- Utilize spectral similarity scores such as the Logarithmic Convoluted Cosine Similarity (LCCS) that account for both frequency and intensity mismatches [10].
Table: Key Research Reagent Solutions for Semi-Empirical Calculations
| Tool/Resource | Function/Purpose | Application Context |
|---|---|---|
| AM1 Method | Geometry optimization and vibrational frequency calculation | IR spectra simulation of organic molecules and 2D materials [3] |
| DFTB3 | Efficient conformational sampling and potential energy surface scanning | Initial conformer search for IR spectrum assignment [10] |
| PM7 | Accurate geometry prediction including noncovalent interactions | Systems requiring improved treatment of hydrogen bonding and dispersion [7] |
| Sparkle/PM7 | Geometry prediction for lanthanide complexes | Coordination compounds and metal-organic frameworks [8] |
| ZINDO | Excited state properties and electronic spectra | UV-Vis spectrum simulation alongside IR studies [3] |
| GFN2-xTB | Generally accurate tight-binding method | Balanced accuracy/efficiency for diverse molecular systems [6] |
The evolution of semi-empirical quantum chemistry methods from MNDO and AM1 to PM6 and PM7 represents a continuous refinement aimed at expanding their applicability while maintaining computational efficiency. For IR spectra simulation, these methods provide valuable tools for researchers, particularly when studying large molecular systems where higher-level calculations remain computationally prohibitive. The choice of method depends critically on the specific application: AM1 maintains utility for IR simulation of organic molecules, DFTB3 offers efficiency for conformational sampling, while PM7 provides improved accuracy for systems where noncovalent interactions are crucial. As semi-empirical methods continue to evolve, their integration with machine learning approaches and fragment-based methods promises to further expand their capabilities for simulating spectroscopic properties of increasingly complex molecular systems.
Comparative Analysis of Key Semi-Empirical Methods
Table 1: Key Characteristics of Semi-Empirical Methods
| Method | Full Name & Developer | Key Theoretical Features | Parameterization Basis | Elements Parameterized |
|---|---|---|---|---|
| AM1 | Austin Model 1 (Dewar et al., 1985) [11] | NDDO approximation; Modified core-core repulsion with Gaussian functions [11] [12] | Atomic spectroscopic data; molecular dipole moments, IPs, geometries [12] | H, C, N, O, F, Al, Si, P, S, Cl, Br, I [11] [13] |
| PM3 | Parametric Method 3 (Stewart, 1989) [14] | Same formalism as AM1; uses two Gaussians for core repulsion [14] | Optimizable parameters fitted to reproduce molecular properties [14] [12] | H, Li, C, N, O, F, Al, Si, P, S, Cl, Br, I & many main-group elements [14] [13] |
| PM6 | Parameterization Method 6 (Stewart, 2007) [15] | NDDO; pairwise interaction corrections to core repulsion; new 1-center integral method for TMs [15] | Pruned experimental data; ab initio/DFT data; dimerization energies [15] | H, C, N, O, common elements, and transition metals [15] |
| PM7 | Parameterization Method 7 (Stewart, 2012) [2] | Rectified NDDO errors; improved non-covalent interactions [2] | Experimental and high-level ab initio data; crystal structures & heats of formation of solids [2] | Broad range, including solids and transition metals [2] |
| GFN-xTB | Geometry, Frequency, and Noncovalent extended Tight-Binding (Grimme group) | Extended tight-binding framework; semi-empirical DFT methods [16] [17] | Fitted to quantum chemical and experimental reference data [16] | Across the periodic table [16] |
Table 2: Performance and Typical Applications
| Method | Performance & Accuracy Notes | Known Limitations | Common Applications |
|---|---|---|---|
| AM1 | Improved heats of formation (MAD ~30 kJ/mol) over MNDO; describes H-bonding (poorly) [12] [18] | Overestimates basicities; incorrect water dimer geometry; strong intermolecular repulsion [12] | Initial geometry optimizations; starting point for forcefield parameterization [11] [12] |
| PM3 | Better H-bonding than AM1; good thermochemical accuracy (MAD ~18 kJ/mol) [12] [18] | Non-physical H-H attractions; overestimates nitrogen inversion barriers [12] | Rapid estimation of molecular properties; large systems [14] [12] |
| PM6 | Better general performance than AM1/PM3; good for geometries and IPs; describes transition metals [15] | Incorrect linear Si-O-H; reduced repulsion for specific atom pairs (e.g., Br-Br) [2] | Biochemical systems; transition metal complexes; large-scale simulations [15] [2] |
| PM7 | Improved ΔHf and geometries for solids; reduced errors vs. PM6 (AUE in ΔHf for solids dropped 60%) [2] | Instabilities with 3rd-row+ elements/transition metals; wavefunction convergence issues [13] | Crystalline solids; biochemical macromolecules; improved reaction barriers (with PM7-TS) [2] |
| GFN-xTB | Moderate performance alone (MAE ~2.5 kcal/mol); highly accurate with DFT single-point correction [16] | Accuracy limits for large, flexible systems; parameterization dependency [16] [17] | Supramolecular assembly; non-covalent complexes; nanoscale systems [16] |
Method Evolution and Workflow Integration
Figure 1: Historical development and relationships between key semi-empirical quantum chemical methods. Solid arrows indicate direct evolutionary lineage within the NDDO family, while the dashed arrow indicates a conceptual influence on the newer GFN-xTB framework.
Computational Protocols for Method Application
Protocol 1: Single-Point Energy and Property Calculation
Purpose: To compute the heat of formation, ionization potential, and dipole moment of a molecule using a semi-empirical method.
Workflow:
- Molecular Structure Input: Provide a 3D molecular structure, either from a database or via a prior geometry optimization.
- Method Selection: Choose the appropriate semi-empirical Hamiltonian (e.g., AM1, PM3, PM6, PM7) in the computational software. Do not specify a basis set, as this is intrinsic to the method [13].
- Calculation Setup:
- Execution: Run the calculation.
- Output Analysis: Extract the final heat of formation (in kcal/mol), ionization potential (from HOMO energy or via ΔSCF), and dipole moment (in Debye) from the output file.
Protocol 2: Geometry Optimization and Frequency Analysis
Purpose: To obtain an optimized molecular geometry and compute vibrational frequencies for IR spectrum simulation.
Workflow:
- Initial Geometry: Start with a reasonable 3D molecular structure.
- Method and Task Selection:
- Select the semi-empirical method.
- Set the task to "Geometry Optimization" followed by "Frequency Calculation."
- Convergence Criteria: Apply standard convergence thresholds for the geometry optimization (e.g., gradient root-mean-square < 0.001 kcal/mol/Å).
- Execution: Run the sequential optimization and frequency jobs.
- Result Validation:
- Check that the optimization converged successfully.
- Confirm the absence of imaginary frequencies for a minimum, or exactly one for a transition state.
- IR Spectrum Generation: Use the computed harmonic frequencies and intensities to simulate the IR spectrum.
Protocol 3: Hybrid GFN-xTB/DFT Protocol for Supramolecular Systems
Purpose: To achieve high accuracy for conformational equilibria and non-covalent interaction energies in large systems at a reduced computational cost [16].
Workflow:
- Geometry Optimization with GFN-xTB: Perform a full geometry optimization of the molecular system (e.g., a host-guest complex or a folded polymer) using the GFN-xTB method.
- Structure Validation: Check the optimized geometry for reasonable bond lengths and angles.
- Single-Point Energy Correction: Using the GFN-xTB-optimized geometry, perform a single-point energy calculation using a more accurate Density Functional Theory (DFT) method, ideally including dispersion correction (e.g., DFT-D3).
- Result Analysis: Use the DFT single-point energy for accurate thermodynamic comparisons (e.g., binding energies, conformational stabilities). This hybrid approach can achieve near-DFT accuracy with a significant reduction in computational time [16].
The Scientist's Toolkit
Table 3: Essential Software and Parameter Resources
| Resource Name | Type | Function | Key Features |
|---|---|---|---|
| MOPAC | Software | The original and a widely used platform for semi-empirical calculations. | Implements AM1, PM3, PM6, PM7; public domain versions available [14] [15]. |
| Gaussian | Software | General-purpose quantum chemistry package. | Includes reimplemented AM1, PM3, PM6, PM7 for analytic gradients and frequencies [13]. |
| GFN-xTB | Software | Standalone program for extended tight-binding calculations. | Efficient for large systems and non-covalent interactions; often used with DFTB+ [16] [17]. |
| GAMESS | Software | Another comprehensive quantum chemistry package. | Supports multiple semi-empirical methods for various chemical systems [14]. |
| Semi-Empirical Parameters | Data | Element-specific parameter sets. | Built-in for standard elements; user-readable for custom parameterization in Gaussian/MOPAC format [13]. |
In the field of computational chemistry, semi-empirical quantum chemical (SQC) methods represent a critical balance between computational cost and quantum mechanical accuracy, making them particularly suitable for studying drug-sized molecules. These methods simplify the complex calculations of ab initio approaches by utilizing empirical parameters and approximations, enabling researchers to handle the large molecular systems and extended time scales common in pharmaceutical research [19] [20]. For research focused on IR spectra simulation, SQC methods provide a computationally feasible pathway to obtaining accurate vibrational frequencies and spectroscopic properties for vast libraries of drug-like compounds, a task that would be prohibitively expensive using purely first-principles calculations [21]. This application note details the quantitative advantages, experimental protocols, and practical implementation of SQC methods for drug discovery applications, with particular emphasis on IR spectral prediction.
Quantitative Efficiency and Accuracy Benchmarks
The computational efficiency of SQC methods does not come without trade-offs in accuracy; however, strategic approaches can mitigate these limitations. The following table summarizes the performance characteristics of various computational methods when applied to drug-like molecules:
Table 1: Performance Comparison of Computational Chemistry Methods for Drug-Sized Molecules
| Method Category | Representative Methods | Computational Speed vs. DFT | Typical System Size Limit | Key Strengths | Key Limitations |
|---|---|---|---|---|---|
| Semi-Empirical (SQC) | PM6, AM1, GFN-xTB | 100 - 1,000x faster [20] | >1,000 atoms [22] | High speed, good for geometry optimization and MD [23] | Lower accuracy, parameter-dependent [19] [20] |
| Neural Network Potentials | ANI, DPA-2-Drug | 1,000 - 10,000x faster [22] | ~70 heavy atoms [22] | Near-DFT accuracy for energies and forces [22] | Requires extensive training data [22] |
| Density Functional Theory (DFT) | PBE, B3LYP, M08-HX | 1x (Benchmark) | ~100 atoms | High accuracy, good for diverse properties [23] | Computationally expensive [22] [23] |
| Ab Initio | MP2, CCSD(T) | 10 - 10,000x slower | <50 atoms | Very high accuracy, gold standard | Extremely computationally expensive [22] |
A key strategy for maintaining both efficiency and accuracy is the hybrid or "modular" approach. Benchmarking studies on quinone-based electroactive compounds demonstrate that performing geometry optimization with a lower-level method (e.g., SQC or force fields) followed by a single-point energy calculation at a higher level of theory (e.g., DFT) can yield results comparable to full high-level optimization at a fraction of the cost [23]. For instance, this approach successfully predicted redox potentials with an RMSE of only ~0.05 V, which is within common experimental error margins [23]. Similarly, a 2025 study on supramolecular assemblies found that applying DFT-level single-point energy corrections to GFN-xTB-optimized geometries reduced the mean absolute error (MAE) from ~5.0 kcal mol⁻¹ to ~1.0 kcal mol⁻¹, achieving "DFT-D3-level accuracy while maintaining a low computational cost" [16].
Application Protocols for Drug Discovery
Protocol 1: High-Throughput Virtual Screening with IR Spectral Prediction
Objective: To rapidly predict the IR spectra of thousands of drug-like molecules from a chemical library for functional group identification or structural fingerprinting.
Workflow:
- Compound Library Preparation: Input molecular structures in SMILES or SDF format. A common source is the ChEMBL database [21].
- Initial Geometry Optimization: Optimize the 3D geometry of each molecule using a fast SQC method such as GFN-xTB or PM6. This step finds the lowest energy conformer.
- Vibrational Frequency Calculation: Using the optimized geometry, calculate the harmonic vibrational frequencies, IR intensities, and Raman activities with the same or a specialized SQC method. The Gaussian software package is commonly used for this step [21].
- Data Compilation & Analysis: Compile the calculated frequencies and intensities into a spectral database. Analyze the spectra to identify characteristic absorption bands or use the data as a fingerprint for machine learning models [21].
Protocol 2: Binding Affinity Estimation via Hybrid Geometry Optimization
Objective: To efficiently estimate the binding affinity of a drug candidate by calculating its interaction energy with a protein binding pocket.
Workflow:
- System Setup: Obtain the 3D structure of the protein-ligand complex, placing the ligand in the binding site.
- Geometry Optimization with SQC: Optimize the geometry of the ligand and key residues in the binding site using an SQC method. This refines the structure at low computational cost.
- High-Level Single-Point Energy Calculation: Perform a single-point energy calculation on the SQC-optimized structure using a more accurate DFT functional (e.g., PBE0 or M08-HX) and an implicit solvation model.
- Energy & Property Analysis: Use the resulting high-level energy to calculate binding energies or other electronic properties relevant to drug action [23].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Software and Datasets for SQC-based IR Spectroscopy Research
| Item Name | Type | Primary Function | Relevance to Research |
|---|---|---|---|
| Gaussian 09/16 | Software Suite | Performs quantum chemical calculations (SQC, DFT, ab initio). | Industry standard for calculating IR/Raman spectra and optimizing molecular geometries [21]. |
| GFN-xTB | Software (SQC Method) | Fast semi-empirical geometry optimization and frequency calculation. | Enables high-throughput screening of molecular structures and vibrational properties with good accuracy [20]. |
| ChEMBL Database | Chemical Database | A manually curated database of bioactive molecules with drug-like properties. | Primary source for structures of drug-like molecules for virtual screening and spectral library generation [21]. |
| ANI/DPA-2-Drug | Neural Network Potential | Machine-learned potential for energy and force prediction. | Provides near-ab initio accuracy at speeds thousands of times faster than DFT for molecular dynamics [22]. |
| Extended Spectral Dataset | Research Dataset | A dataset of computed Raman and IR spectra for molecules from ChEMBL. | Serves as a training and benchmarking resource for machine learning models predicting spectra from structures [21]. |
Semi-empirical quantum chemical methods provide an indispensable toolset for computational drug discovery, particularly for IR spectra simulation and other applications involving drug-sized molecules. Their primary advantage lies in a favorable balance of computational efficiency and acceptable accuracy, enabling studies at a scale that is impractical for pure ab initio methods. By integrating these methods into hierarchical workflows—such as using SQC for initial geometry sampling and DFT for final energy calculations—researchers can maximize both throughput and predictive power. As machine learning potentials continue to evolve, the boundaries of speed and accuracy in computational spectroscopy and drug design are expected to be pushed even further.
Semi-empirical methods for infrared (IR) spectra simulation represent a critical tool for researchers seeking to correlate molecular structures with experimental spectroscopic data. These methods fill an important niche between highly accurate but computationally expensive ab initio quantum mechanical approaches and fast but often insufficiently precise classical force fields. Their value is particularly evident in high-throughput screening, large biomolecular systems, and automated discovery platforms where computational efficiency is paramount. However, the strategic application of these methods requires a thorough understanding of their inherent limitations, accuracy trade-offs, and systematic error sources. This Application Note provides a structured analysis of these critical limitations and offers detailed protocols for their identification, quantification, and mitigation within research and development workflows, particularly for drug development professionals who rely on computational spectroscopy for molecular characterization.
Accuracy Trade-offs: A Quantitative Analysis
The computational efficiency of semi-empirical methods is achieved through approximations and parameterization that inevitably introduce specific, quantifiable errors in simulated IR spectra. Understanding these trade-offs is essential for selecting appropriate methods and interpreting results with necessary caution.
Table 1: Quantitative Accuracy Benchmarks of Computational Methods for IR Spectra Simulation
| Computational Method | Computational Speed (Relative to DFT) | Typical Frequency Error (cm⁻¹) | Key Limitations & Systematic Errors |
|---|---|---|---|
| Semi-empirical (e.g., AM1) [3] | ~10²-10⁴ faster | Can exceed 10% for specific vibrational modes [3] | Poor description of anharmonic effects; inaccurate for hydrogen-bonding and long-range interactions; parameter-dependent errors. |
| Density Functional Theory (DFT) [10] | 1x (Baseline) | 10-30 (with appropriate basis sets) [10] | Still significant for precise vibrational assignment; functional-dependent performance for dispersion forces. |
| AI-Enhanced Quantum Mechanics (e.g., AIQM1/2) [24] [25] [26] | ~10-100x faster than DFT [26] | Approaching DFT accuracy (specific benchmarks not fully established) [24] | Performance can degrade on molecules outside training data; requires careful validation for novel chemical structures. |
| GFN2-xTB (Semi-empirical) [26] | ~10³ faster than DFT [26] | Data not explicitly provided in search results | Generally less accurate than DFT for vibrational frequencies; known challenges with specific element types. |
The data in Table 1 illustrates the fundamental trade-off between computational speed and accuracy. Semi-empirical methods like AM1, while highly efficient, can exhibit errors exceeding 10% for certain vibrational modes, necessitating careful validation against experimental data [3]. The parameterization of these methods, often derived from a limited set of experimental data or higher-level calculations, is a primary source of systematic error, making them less reliable for molecules or functional groups far from their parameterization domain.
Systematic errors in semi-empirical IR simulations arise from foundational approximations in the theoretical model. The following table categorizes key sources and suggests mitigation strategies.
Table 2: Systematic Error Sources in Semi-Empirical IR Simulations and Mitigation Protocols
| Error Source | Impact on Simulated IR Spectrum | Recommended Mitigation Strategies |
|---|---|---|
| Incomplete Basis Set | Inaccurate harmonic force constants, leading to frequency shifts, especially for polarizable bonds. [10] | Use methods with polarization functions where feasible. [10] For pure semi-empirical methods, validate against a higher-level method (e.g., DFT) for a representative subset. |
| Neglect of Anharmonicity | Inaccurate overtone and combination band positions; incorrect bandwidth and intensity estimation. [27] | Apply empirical scaling factors (though this is a correction, not a solution). For critical applications, use molecular dynamics approaches or higher-level methods that account for anharmonicity. [27] |
| Approximate Treatment of Electron Correlation | Systematic errors in binding energies and potential energy surfaces, affecting frequencies for weakly interacting systems (e.g., van der Waals complexes). [28] | Employ dispersion-corrected semi-empirical methods or Δ-learning approaches like AIQM2 that add ML-based corrections to a semi-empirical baseline. [26] |
| Conformational Sampling Inadequacy | Spectrum represents a single, low-energy conformation rather than a thermally averaged ensemble, missing broadening and key bands. [27] | Perform conformational searching and generate spectra as an ensemble average from multiple low-energy conformers using MD simulations. [27] |
| Parameterization Domain Mismatch | Poor performance and large errors for molecules, functional groups, or elements not well-represented in the training/parameterization set. | Validate method performance on known reference molecules similar to the target system before application. Use more universal methods like GFN2-xTB or AI-enhanced models. [26] |
Experimental Protocols
Protocol 1: Benchmarking and Validating Semi-Empirical Methods
This protocol outlines a standardized workflow for evaluating the accuracy of a semi-empirical method for a specific chemical system of interest, ensuring its reliable application in research.
1. Reference Data Acquisition:
- Obtain high-quality experimental IR spectra for one or more well-characterized reference molecules structurally similar to your target system. Record experimental conditions (solvent, temperature, concentration).
- Reagent Solution: Use standard reference compounds from suppliers like Sigma-Aldrich or TCI. For aqueous systems, N-methylacetamide (NMA) is a prototypical model for peptide bonds [27].
2. Computational Model Generation:
- Generate an initial 3D molecular structure for the reference molecule.
- Perform geometry optimization and vibrational frequency calculation using the semi-empirical method under investigation (e.g., AM1, GFN2-xTB). Most semi-empirical packages use a second-order Taylor expansion of energy to optimize geometry and calculate the Hessian matrix for vibrational frequencies [3].
- Computational Note: The convergence limit for geometry optimization is typically set to a gradient of 0.05 kcal/mol/Ångstrom to ensure a stable structure before force calculation [3].
3. Spectral Comparison and Error Quantification:
- Compare the calculated harmonic frequencies with experimental peak positions.
- Calculate the mean absolute error (MAE) and root mean square error (RMSE) for all major vibrational bands.
- A suitable method should typically predict frequencies within 10% of experimental values for the vibrational modes of interest [3]. If errors exceed this threshold, consider using a different method or applying a systematic scaling factor.
4. Method Selection/Correction:
- Based on the error analysis, decide whether the semi-empirical method is fit for purpose.
- If systematic errors are consistent, derive a linear scaling factor for frequency correction.
- Alternatively, transition to a more accurate method, such as a hybrid DFT functional (e.g., B3LYP-D3(BJ)) with a polarized basis set for final production calculations [10] [28].
Protocol 2: Workflow for IR Spectra Simulation of a Novel Molecule
This protocol describes the end-to-end process for simulating and interpreting the IR spectrum of a novel molecular entity, integrating steps to manage accuracy trade-offs.
1. System Preparation and Conformational Sampling:
- Structure Building: Construct an initial 3D model of the novel molecule.
- Conformational Search: Use a fast method (e.g., molecular mechanics or semi-empirical like DFTB3) to scan the potential energy surface and identify low-energy conformers [10]. Retain all unique conformers within a ~15 kJ/mol energy window.
- Reagent Solution: Software tools like MAXIMOBY or GROMACS can be used for initial system preparation and solvation, placing the molecule in a cubic box with explicit solvent molecules (e.g., TIP4P water) [27].
2. Geometry Optimization and Frequency Calculation:
- For each low-energy conformer, perform a geometry optimization using the selected semi-empirical method (e.g., AM1, GFN2-xTB) or a higher-level method if resources allow.
- Critical Step: Calculate the vibrational frequencies and IR intensities from the optimized geometry. This is typically done by diagonalizing the mass-weighted Hessian (force constant) matrix to obtain normal modes and their frequencies [3] [27].
3. Ensemble Spectrum Generation:
- Generate a composite spectrum by averaging the spectra of all low-energy conformers, weighted by their Boltzmann population at the target temperature (e.g., 298 K). This accounts for conformational flexibility and provides a more realistic spectrum comparable to experiment [27].
4. Interpretation and Validation:
- Assign calculated vibrational bands to specific molecular motions (e.g., C=O stretch, N-H bend) by visualizing the normal modes.
- If possible, compare the overall spectral profile with any available experimental data or with spectra of analogous compounds predicted at a higher level of theory (e.g., DFT) to check for major discrepancies.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Semi-Empirical IR Spectroscopy
| Tool / Resource | Function | Application Note |
|---|---|---|
| Semi-empirical Methods (AM1, GFN2-xTB) | Fast geometry optimization and vibrational frequency calculation. | Ideal for initial screening and large systems. Requires validation. [3] |
| DFT Methods (e.g., B3LYP-D3(BJ)) | Higher-accuracy reference calculations for benchmarking. | Use with polarized basis sets for reliable frequency prediction. [10] [28] |
| AI-Enhanced Models (AIQM1/2) | Accelerated calculations targeting coupled-cluster accuracy. | Implemented in MLatom; promising for balancing speed and accuracy. [24] [25] [26] |
| Conformational Search Software (GROMACS, MAXIMOBY) | Generates thermally accessible structural ensembles. | Critical for simulating realistic, thermally averaged spectra in solution. [27] |
| Vibrational Analysis & Visualization Software | Assigns vibrational modes to spectral peaks by animating molecular vibrations. | Essential for interpreting spectra and identifying systematic errors in specific bond types. |
Practical Implementation: Workflows and Biomedical Applications
Within computational chemistry, semi-empirical quantum chemistry methods offer a unique balance between computational cost and electronic structure detail, positioning them as indispensable tools for studying large molecular systems such as those found in drug development [29] [30]. These methods are approximate versions of the Hartree-Fock formalism where specific integrals are omitted or replaced with empirical parameters, resulting in a significant reduction in computational expense [29] [30]. The primary strength of semi-empirical methods lies in their ability to perform rapid geometry optimizations and vibrational frequency calculations for very large molecules, tasks that would be prohibitively expensive using higher-level ab initio or density functional theory (DFT) methods [29] [3]. This Application Note details a standardized protocol for employing semi-empirical methods to compute infrared (IR) spectra, from initial geometry optimization to the final frequency analysis, providing a robust framework for researchers aiming to interpret the vibrational characteristics of complex molecular systems.
Theoretical Background of Semi-Empirical Methods
Semi-empirical methods are rooted in the Hartree-Fock formalism but incorporate drastic simplifications to achieve computational efficiency. A key approximation is the Neglect of Diatomic Differential Overlap (NDDO), which significantly reduces the complexity and number of two-electron integrals that must be computed [30] [2]. The parameters introduced by these approximations are then fitted against experimental data or high-level ab initio results, enabling the methods to partially account for electron correlation effects at a minimal computational cost [30].
Several families of semi-empirical methods have been developed over the years, each with specific parameterizations and target applications. The most common methods, often implemented in packages like MOPAC, AMPAC, and SPARTAN, include MNDO, AM1, and PM3, and their more modern successors PM6 and PM7 [30] [2]. These are primarily parameterized to reproduce experimental heats of formation, dipole moments, ionization potentials, and molecular geometries [30] [2]. For specialized applications such as simulating electronic spectra, methods like ZINDO are preferred [30] [3]. Furthermore, modern semi-empirical approaches like the GFNn-xTB (Geometry, Frequency, Noncovalent, extended Tight Binding) family offer particularly robust performance for geometry optimization, vibrational frequency calculation, and the treatment of non-covalent interactions in large molecules [30] [31].
A notable alternative that bridges the gap between semi-empirical and minimal-basis ab initio methods is HF-3c. This is Grimme's Hartree-Fock method corrected with three additive terms: a dispersion correction (D3), a basis set superposition error correction (gCP), and a short-range basis set incompleteness correction. While computationally more intensive than traditional semi-empirical methods, HF-3c offers significantly improved robustness and reliability [29].
Standard Computational Workflow: A Step-by-Step Protocol
The following section outlines a detailed, step-by-step protocol for conducting geometry optimization and subsequent frequency calculations using semi-empirical methods. Adherence to this workflow is critical for generating physically meaningful and reliable IR spectra.
The diagram below illustrates the logical sequence of the entire computational process, from initial structure preparation to the final analysis of the IR spectrum.
Detailed Experimental Protocol
Step 1: Initial Structure Preparation
- Procedure: Construct or obtain the initial 3D molecular structure. This can be done using chemical drawing software or by modifying a structure from a database. Ensure the structure is chemically reasonable.
- Critical Parameters:
- Molecular Charge: Set the total charge of the system (e.g., 0 for neutral molecules).
- Spin Multiplicity: Define the correct spin multiplicity (e.g., 1 for singlet closed-shell systems).
Step 2: Geometry Optimization
- Objective: Find the nearest local minimum on the potential energy surface, corresponding to a stable molecular conformation.
- Input Settings:
- Method Selection: Choose an appropriate semi-empirical method. For general organic molecules, PM7 is recommended due to its improved treatment of non-covalent interactions and good overall accuracy compared to its predecessors [2]. For very large systems or where non-covalent interactions are critical, GFN2-xTB is an excellent choice [31].
- Convergence Criteria: Set stringent convergence thresholds for the geometry optimization. A common and effective criterion is to require the energy gradient to be below 0.05 kcal/mol/Ångstrom [3]. Powerful gradient-based minimization algorithms, which are standard in modern codes, should be used to efficiently conserve molecular symmetry during optimization [32].
- Execution: Run the optimization job using a computational chemistry package such as ORCA, Gaussian, MOPAC, or xtb.
Step 3: Convergence Verification
- Procedure: After the optimization job completes, carefully inspect the output log file.
- Success Criteria: The calculation must explicitly state that it has converged and the final energy gradient should be below the specified threshold. An unconverged optimization will lead to invalid results in the subsequent frequency calculation.
Step 4: Frequency Calculation
- Objective: Compute the second derivatives of the energy (the Hessian matrix) at the optimized geometry to obtain the vibrational frequencies and IR intensities.
- Input Settings:
- Geometry: Use the optimized geometry coordinates from Step 2.
- Method: It is critical to use the exact same semi-empirical method and parameters (e.g., PM7) as were used for the geometry optimization. Failure to do so makes the frequency calculation meaningless.
- Keyword: Use the appropriate keyword to request a frequency calculation (e.g.,
Freqin Gaussian and ORCA).
- Execution: Run the frequency calculation job.
Step 5: Analysis and Validation of Results
- Validation - Absence of Imaginary Frequencies: A successful frequency calculation at a true energy minimum must yield zero imaginary frequencies. The presence of one or more imaginary frequencies indicates that the geometry is not at a minimum but rather at a saddle point (e.g., a transition state), and the geometry optimization must be restarted, potentially from a different initial structure.
- IR Spectrum Interpretation:
- The output provides a list of vibrational frequencies (in cm⁻¹) and their corresponding IR intensities.
- These data can be plotted to generate a simulated IR spectrum, which can be compared directly with experimental data.
- The vibrational normal modes can be visualized to assign specific peaks to molecular motions (e.g., O-H stretch, C=O stretch) [3] [33].
Essential Computational Tools and Reagents
The following table summarizes key software and methodological "reagents" required to execute the described workflow successfully.
Table 1: Research Reagent Solutions for Semi-Empirical IR Simulation
| Item Name | Function/Description | Example Usage in Workflow |
|---|---|---|
| PM7 Hamiltonian | A modern semi-empirical method parameterized for improved accuracy for geometries and heats of formation of organic molecules and solids [2] [13]. | Default method for geometry optimization and frequency calculation of organic drug-like molecules. |
| GFN2-xTB | A highly efficient semi-empirical tight-binding method well-suited for large systems, providing good geometries and vibrational frequencies [31]. | Primary method for very large molecules (e.g., protein-ligand complexes) or when computational speed is paramount. |
| ZINDO Method | A semi-empirical method specially parameterized for simulating electronic excitation spectra and UV-Vis properties [30] [3]. | Not for IR spectra, but used in a separate single-point calculation to predict UV-Vis spectra after obtaining the optimized geometry. |
| HF-3c Method | A cost-effective minimal-basis-set Hartree-Fock method with semi-empirical corrections for dispersion (D3) and basis set effects (gCP) [29]. | An attractive, more robust alternative to traditional semi-empirical methods when higher accuracy is needed. |
| ORCA Software | A widely used quantum chemistry package with comprehensive support for semi-empirical methods (MNDO, AM1, PM3, PM6, HF-3c) and the xtb interface [29] [34]. | General-purpose platform for running the entire workflow. Input: ! PM7 Opt followed by ! PM7 Freq. |
| xtb Program | A dedicated software for fast semi-empirical calculations using the GFNn-xTB family of methods [31]. | Specialized for high-throughput geometry optimizations and frequency calculations of large systems. Command: xtb --opt --gfn 2 |
| Force Calculation | The computational step following optimization that calculates the Hessian matrix (force constants) to determine vibrational frequencies [3]. | This is the core step of the frequency job, activated by the Freq keyword in input files. |
Performance and Application Considerations
Quantitative Performance of Methods
The choice of semi-empirical method significantly impacts the accuracy of the computed geometries and vibrational frequencies. The following table summarizes typical performance characteristics based on benchmark studies.
Table 2: Performance Comparison of Common Semi-Empirical Methods
| Method | Typical Error in Bond Lengths (Å) | Typical Error in ΔHf (kcal/mol) | Strengths and Preferred Applications |
|---|---|---|---|
| PM3 | ~0.02 | ~8.0 | Historically widely used; largely superseded by PM6 and PM7. |
| PM6 | ~0.015 | ~6.3 | Improved accuracy over PM3/AM1; known for some specific faults (e.g., linear Si-O-H) [2]. |
| PM7 | ~0.014 | ~5.7 | Reduced errors for solids and non-covalent interactions; recommended for general use [2]. |
| GFN2-xTB | Varies | Varies | Excellent for non-covalent interactions, large systems, and vibrational frequencies; very fast [31]. |
| AIQM1 | N/A | N/A | A new AI-based model targeting CCSD(T) accuracy for IR spectra, close to DFT quality at semi-empirical cost [33]. |
Protocol for IR Spectra of 2D Materials
The analysis of functional groups on 2D materials like graphene quantum dots (GQDs) presents a unique challenge, as IR spectra traditionally assume 3D materials. A specialized protocol has been demonstrated using pyrene-like molecules as models for GQDs [3]:
- Model Construction: Build a molecular model representing a fragment of the 2D material (e.g., a pyrene-like GQD, pGQD).
- Geometry Optimization and IR Simulation with AM1: Perform geometry optimization and subsequent vibrational frequency calculation using the AM1 semi-empirical method. The AM1 method has proven successful in simulating the vibrational spectrum of organic materials and agrees well with experimental results for these systems [3].
- Spectral Analysis: Focus the analysis on key regions of the IR spectrum:
- 1400–2000 cm⁻¹: For the C=C stretching mode of the aromatic backbone.
- 3000–3500 cm⁻¹: For C–H and O–H stretching vibrations from functional groups.
- This approach allows for the sensitive identification of functionalization at the edge versus the center of the 2D surface, which is difficult with standard experimental analysis alone [3].
Troubleshooting and Best Practices
- Imaginary Frequencies: If one or two small imaginary frequencies (< 50i cm⁻¹) appear, they can sometimes be ignored as numerical artifacts. However, larger or numerous imaginary frequencies indicate a failed optimization. Solution: Restart the geometry optimization with a different initial guess or a different optimization algorithm.
- Wavefunction Instabilities (PM7): For systems with third-row and higher elements, the PM7 wavefunction can sometimes be unstable. Solution: Try an alternate SCF algorithm (e.g.,
SCF=YQCin Gaussian) or use a different initial guess for the density matrix [13]. - Accuracy Limitations: Semi-empirical methods are parameterized for specific element types and molecular environments. Their accuracy can be poor for systems outside their parameterization set. Solution: For critical results, validate findings by single-point energy calculations or geometry optimizations on key structures using a higher-level method like DFT.
- Anharmonic Effects: Standard frequency calculations are performed within the harmonic approximation, which can cause inaccuracies for X-H stretches and other strongly anharmonic modes. Solution: For more accurate spectra, especially at high temperatures, consider using ab initio molecular dynamics (AIMD) approaches with semi-empirical methods (e.g., GFN2-xTB in PyRAMD software) to account for anharmonicity [34].
Graphene Quantum Dots (GQDs) represent a zero-dimensional class of nanomaterials that have garnered significant scientific interest due to their exceptional physicochemical properties, which include quantum confinement, edge effects, tunable photoluminescence, high surface-to-volume ratio, and excellent biocompatibility [35] [36]. The functionalization of GQDs with specific functional groups is a critical determinant of their optical, electronic, and chemical behavior, directly influencing their performance in applications ranging from biosensing and bioimaging to drug delivery and energy storage [37] [38]. Unlike three-dimensional (3D) materials, the identification and analysis of functional groups in two-dimensional (2D) materials like GQDs present unique challenges, as traditional Infrared (IR) spectroscopy often assumes 3D sample geometries, making it difficult to distinguish between functionalizations located at the edges versus the center of the 2D surface [3].
Within the context of a broader thesis on semi-empirical methods for IR spectra simulation, this application note provides a detailed protocol for the identification of functional groups in GQDs. We emphasize the utility of semi-empirical computational methods, such as the Austin Model 1 (AM1), which serve as a powerful auxiliary tool for interpreting experimental Fourier-Transform Infrared (FTIR) spectra. These methods enable researchers to correlate complex vibrational modes with specific molecular structures, thereby facilitating a more accurate analysis of functionalized GQDs [3]. This document is structured to guide researchers and drug development professionals through the fundamental principles, computational and experimental methodologies, and practical applications of functionalized GQDs in the biomedical field.
Fundamental Properties and Functionalization of GQDs
Structural and Optical Characteristics
GQDs are typically defined as single or few-layer graphene fragments with lateral dimensions less than 100 nm, featuring a honeycomb lattice of sp²-hybridized carbon atoms [39] [36]. Their nano-scale size induces quantum confinement effects, which endows them with a tunable band gap—a property absent in pristine graphene. This tunability is directly manifested in their size-dependent and surface-state-influenced photoluminescence (PL) [40] [36]. The optical properties of GQDs are primarily governed by two factors: the conjugated π-domain within the core, which can be influenced by the size and shape of the dot, and the surface/edge states, which are determined by the attached functional groups and the type of edge structure (e.g., zigzag vs. armchair) [37] [38]. For instance, functional groups can introduce intermediate energy levels within the bandgap, leading to shifts in absorption and emission spectra, such as red-shifts observed upon conjugation with drug molecules like doxorubicin (DOX) [38].
Common Functional Groups and Their Impact
The surface and edges of GQDs are often decorated with various oxygen-containing functional groups, such as hydroxyl (-OH), carboxyl (-COOH), carbonyl (C=O), and epoxy groups, when synthesized from precursor materials like graphene oxide [40] [35]. These groups significantly enhance the hydrophilicity, colloidal stability, and biocompatibility of GQDs, making them suitable for aqueous biological applications [35]. Furthermore, intentional heteroatom doping—the incorporation of elements like nitrogen (N), sulfur (S), or boron (B) into the carbon lattice—is a prevalent strategy to perturb the local electronic environment and tailor the optical and electrochemical properties of GQDs [37] [35]. Nitrogen doping, for example, has been shown to enhance fluorescence quantum yield and introduce new reaction sites for further chemical conjugation [37].
Table 1: Common Functional Groups in GQDs and Their Spectral and Functional Impacts.
| Functional Group/Dopant | Characteristic IR Vibrations (cm⁻¹) | Key Impacts on GQD Properties |
|---|---|---|
| Hydroxyl (-OH) | 3000-3500 (broad, stretching) [3] | Enhances hydrophilicity and biocompatibility; serves as a site for covalent conjugation [35]. |
| Carbonyl (C=O) | ~1700 (stretching) [3] | Increases chemical reactivity; involved in charge transfer processes [37]. |
| Aromatic C=C | 1400-2000 (skeletal stretching) [3] | Constitutes the core sp² carbon network; responsible for intrinsic fluorescence [3]. |
| C-O-C (Epoxy/ether) | 1220-1290 (stretching) [3] | Can create structural defects; influences electronic structure [3]. |
| Nitrogen Dopant (N) | N/A | Alters electronic density; creates new energy levels; enhances photoluminescence quantum yield and catalytic activity [37] [35]. |
Computational Protocol: Semi-Empirical IR Simulation
Semi-empirical methods provide a balanced approach between computational cost and accuracy, making them ideal for simulating the IR spectra of large molecular systems like GQDs. The following protocol outlines the steps for using the AM1 method to simulate and interpret the IR spectra of functionalized GQDs.
Computational Methodology
The simulation process involves several key stages, from model construction to spectral analysis, as validated in studies using pyrene-like molecules as models for GQDs [3].
Molecular Model Construction:
- Begin by constructing a molecular model representing the GQD structure. Polycyclic aromatic hydrocarbons (PAHs) like pyrene (C₁₆H₁₀) or larger analogs (e.g., 9-ring or 16-ring structures) serve as excellent initial models for the GQD core [3].
- Functionalize the model by adding relevant functional groups (e.g., -OH, -COOH) or heteroatoms (e.g., N) at specific positions—either on the edges or at the center of the molecular surface—to mimic doped or functionalized GQDs.
Geometry Optimization:
- Perform initial geometry optimization using molecular mechanics to obtain a reasonable starting structure.
- Refine the geometry using a semi-empirical method, specifically the AM1 Hamiltonian. The optimization should run until the gradient of the energy change converges below a threshold of 0.05 kcal/mol/Ångstrom, ensuring a stable, minimum-energy structure [3].
Vibrational Frequency Calculation:
- Using the optimized geometry, conduct a force calculation to determine the vibrational modes (frequencies and intensities) of the molecule.
- The output will be a simulated IR spectrum. It is critical to validate the method by comparing the calculated frequencies for a known molecule (e.g., benzene) with experimental data. The semi-empirical technique is deemed suitable if the predicted frequencies are within 10% of the experimental values [3].
Spectral Analysis and Functional Group Assignment:
- Analyze the simulated spectrum by focusing on key vibrational regions:
- Compare the spectra of pristine and functionalized models. The appearance of new peaks or shifts in existing peaks upon functionalization directly indicates the presence of new chemical bonds and allows for the assignment of functional groups. This approach can successfully distinguish between edge and center-surface functionalization [3].
The following workflow diagram illustrates the key steps in this computational protocol:
Figure 1: Workflow for Semi-Empirical IR Simulation of GQDs.
Experimental Validation Protocol
While computational simulation provides predictive insights, experimental validation is essential for confirming the presence of specific functional groups. This protocol details the use of FTIR spectroscopy for this purpose.
Sample Preparation and FTIR Measurement
- Synthesis of GQDs: GQDs can be synthesized via top-down or bottom-up approaches. A common bottom-up method is the thermal pyrolysis of small organic molecules like citric acid. In a typical procedure, citric acid is heated at a defined temperature (e.g., 200°C) until it liquefies and subsequently solidifies, forming GQDs [35]. For doped GQDs, precursors like urea can be added to the citric acid before pyrolysis to incorporate nitrogen atoms [35].
- FTIR Characterization:
- Prepare a sample pellet by mixing a small amount of the synthesized GQD powder with potassium bromide (KBr).
- Acquire the FTIR spectrum in the transmission mode, scanning across a wavenumber range of 4000-400 cm⁻¹.
- Spectral Interpretation:
- Identify the characteristic absorption peaks and correlate them with specific vibrational modes, as outlined in Table 1.
- Directly compare the experimental FTIR spectrum with the semi-empirically simulated spectrum. The computational model aids in deconvoluting overlapping peaks and assigning complex vibrational modes to specific functionalization sites (edge vs. center) on the GQD surface [3].
Applications in Drug Development
The precise identification and engineering of functional groups on GQDs are pivotal for advancing their biomedical applications. Functionalized GQDs serve as versatile platforms in drug delivery and bioimaging.
Drug Delivery Systems: The large surface area and π-π conjugation of GQDs enable high drug-loading capacity. Functional groups facilitate the conjugation of therapeutic agents. For instance, the anticancer drug Doxorubicin (DOX) can be covalently linked to GQDs via C-O, C-N, or C-C bonds. Theoretical studies using Density Functional Theory (DFT) indicate that such covalent conjugation leads to a red-shift in the absorption and emission spectra of the GQDs, a critical feature for tracking the drug carrier. Furthermore, these systems can exhibit strong near-infrared photoluminescence, which is beneficial for deep-tissue imaging [38]. Surface functional groups also allow for the grafting of targeting ligands and the design of pH-responsive drug release systems, minimizing off-target effects [35] [38].
Bioimaging and Biosensing: The tunable photoluminescence of GQDs, achieved through surface functionalization and heteroatom doping, makes them excellent contrast agents for bioimaging. Their low cytotoxicity and high biocompatibility are significant advantages over heavy-metal-based quantum dots [35] [39]. Functional groups enable the development of highly sensitive fluorescence-based biosensors. For example, GQDs can be engineered with specific receptors for glucose, allowing for the development of continuous monitoring systems for diabetes management [39]. The sensing mechanism often relies on fluorescence "turn-off" or "turn-on" responses upon interaction with the target analyte [37].
The following diagram illustrates the multi-functional role of a functionalized GQD in a theranostic (therapy + diagnostic) application:
Figure 2: Theranostic Applications of Functionalized GQDs.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagents and Materials for GQD Functionalization and Analysis.
| Reagent/Material | Function/Application | Brief Explanation |
|---|---|---|
| Citric Acid | Carbon precursor for bottom-up synthesis [35]. | Serves as a common, low-cost source of carbon atoms; upon thermal decomposition, it forms the sp² carbon network of GQDs. |
| Urea | Nitrogen dopant precursor [35]. | Provides a source of nitrogen atoms during synthesis, which become incorporated into the GQD lattice, enhancing fluorescence and electronic properties. |
| Graphite Oxide / Graphene Oxide | Precursor for top-down synthesis [40]. | The oxidized form of graphite, containing abundant oxygen functional groups, can be exfoliated and broken down into nano-sized GQDs. |
| Potassium Bromide (KBr) | Medium for FTIR sample preparation [3]. | A transparent IR material used to create pellets for transmission-mode FTIR analysis of solid GQD samples. |
| Doxorubicin (DOX) | Model chemotherapeutic drug [38]. | Used in research to study GQD-based drug delivery systems; its conjugation to GQDs and release kinetics are extensively investigated. |
| Boric Acid / Sulfur-containing compounds | Dopant precursors for other elements [35]. | Used as sources for boron (B) or sulfur (S) to create heteroatom-doped GQDs with tailored properties. |
The investigation of metal-drug interactions is a critical frontier in modern pharmaceutical science, particularly concerning Schiff base complexes. These compounds, characterized by an azomethine group (-C=N-), are biologically favored platforms with outstanding pharmaceutical importance due to the fabrication of ligands and stable metal complexes [41]. The integration of semi-empirical computational methods for Infrared (IR) spectra simulation provides a powerful toolkit for predicting the structural and vibrational characteristics of these complexes prior to synthetic efforts, thereby accelerating the drug discovery pipeline [3] [42]. This document outlines detailed application notes and experimental protocols for the characterization of pharmaceuticaly relevant Schiff base metal complexes, framed within a broader thesis on semi-empirical IR spectroscopy.
Theoretical Foundation: Semi-Empirical IR Spectroscopy
Semi-empirical quantum chemistry methods offer a balanced approach, providing faster computational speeds than ab initio methods while maintaining a high degree of accuracy for vibrational frequency calculations, making them ideal for the analysis of complex molecular structures like Schiff bases and their metal complexes [3].
- Method Selection and Validation: Among the various semi-empirical methods, the Austin Model 1 (AM1) has demonstrated superior performance in simulating IR spectra for organic and coordination compounds. Studies on aromatic amines and pyrene-like molecules have shown that AM1 provides the best linearity between calculated and experimental frequencies, with a correlation coefficient (cc) of 0.9993 [42]. This makes it particularly suitable for identifying functional groups in complex 2D material-like structures often formed by large Schiff base complexes [3].
- Spectral Analysis Focus: For Schiff base complexes, IR analysis should focus on key vibrational modes:
- Electronic Transitions: The ZINDO method (Zerner's Intermediate Neglect of Differential Overlap) is highly effective for calculating excitation spectra and simulating electronic transitions in the ultraviolet-visible (UV-Vis) region (200–900 nm). This is crucial for understanding the fluorescence properties of Schiff base probes used in bioimaging [3] [43].
Application Notes: Pharmacological Activities of Schiff Base Complexes
Schiff bases and their metal complexes exhibit a broad spectrum of biological activities. The quantitative data from recent studies (2001-2024) are summarized in the tables below for easy comparison [41].
Table 1: Antibacterial and Antifungal Activity of Schiff Base Metal Complexes [41]
| Activity Type | Test Organisms | Reported Efficacy (Least Inhibitory Concentration) | Key Metal Ions |
|---|---|---|---|
| Antibacterial | Gram-positive (S. aureus, S. pyogenes, B. anthracis) | Variable MIC values | Cu, Co, Ni, Zn, Hg, Cd |
| Gram-negative (E. coli, P. aeruginosa, S. typhi) | Variable MIC values | Cu, Co, Ni, Zn, Hg, Cd | |
| Antifungal | A. niger, C. albicans, A. flavus | Variable zone of inhibition / MIC | Cu, Ni, Zn, Co, Hg, Ce |
Table 2: Antiviral and Anticancer Activity of Schiff Base Complexes [41] [43]
| Activity Type | Test Model / Cell Line | Reported Efficacy (IC₅₀ values) | Key Metal Ions |
|---|---|---|---|
| Antiviral | HIV, RSV, HSV, Coronavirus | In vitro inhibitory effects | Cu, Pt, Pd, Co, Fe |
| Anticancer | PC-3, HeLa, HepG-2, MCF-7, HEK-293, HCT-116 | Cytotoxic effects, induction of apoptosis | Cu, Pt, Pd, Zn, Fe, Co |
Case Study: Cu(II) Schiff Base Complexes in Anticancer Therapy and Imaging
Copper(II) complexes have drawn significant attention due to their distinct structural features and potent biological activities [43]. These complexes often exhibit enhanced anticancer activity compared to the free Schiff base ligands. Their proposed mechanisms of action include:
- DNA Binding and Damage: Interaction with DNA via intercalation or groove binding, leading to the disruption of replication and transcription [43].
- Reactive Oxygen Species (ROS) Generation: Catalyzing the production of ROS, which causes oxidative stress and triggers apoptosis in cancer cells [43].
- Enzyme Inhibition: Interfering with the function of key enzymes, such as topoisomerases, crucial for cancer cell proliferation [41].
Furthermore, Schiff base derivatives function as excellent platforms for fluorescent probes. Their electronic structure changes upon binding to metal ions like Cu²⁺, resulting in variations in fluorescence intensity and wavelength. This allows for the sensitive detection and quantification of metal ions in biological samples and enables real-time monitoring of their distribution within live cells, a technique vital for cancer research [43].
Experimental Protocols
Protocol 1: Synthesis of a Schiff Base Ligand and its Cu(II) Complex
Aim: To synthesize and characterize a model N,O-donor Schiff base ligand and its corresponding Cu(II) complex for cytotoxic evaluation [41] [43].
Workflow:
Materials:
- Reagents: Salicylaldehyde derivative, 2-aminophenol derivative, copper(II) chloride/acetate, absolute ethanol, methanol.
- Equipment: Round-bottom flask, condenser, magnetic stirrer with hotplate, Buchner funnel, vacuum pump.
Procedure:
- Ligand Synthesis: Dissolve the aldehyde (1 mmol) and primary amine (1 mmol) in 20 mL of absolute ethanol in a 50 mL round-bottom flask. Add 2-3 drops of glacial acetic acid as a catalyst. Attach a reflux condenser and heat the mixture with stirring at 70-80°C for 4-6 hours.
- Ligand Isolation: Allow the reaction mixture to cool to room temperature. Precipitate the crude product by partial evaporation or addition of cold water. Collect the solid using vacuum filtration and wash with cold ethanol. Recrystallize from a suitable solvent (e.g., ethanol or methanol) to obtain pure Schiff base ligand.
- Metal Complexation: Dissolve the purified Schiff base ligand (1 mmol) in 15 mL of warm methanol. In a separate beaker, dissolve the Cu(II) salt (0.5-1 mmol) in 5 mL of distilled water.
- Complex Formation: Add the aqueous metal salt solution dropwise to the methanolic ligand solution with constant stirring. A color change and/or precipitate formation is typically observed.
- Complex Isolation: Continue stirring for 1-2 hours at room temperature. Filter the precipitated complex, wash successively with water, cold methanol, and diethyl ether, and dry in a desiccator.
Protocol 2: Computational IR Spectrum Simulation using AM1
Aim: To simulate and analyze the IR spectrum of the synthesized Schiff base ligand using the AM1 semi-empirical method [3] [42].
Workflow:
Materials:
- Software: Computational chemistry package capable of semi-empirical calculations (e.g., HyperChem, Winmostar, ORCA, Gaussian).
- Hardware: Standard computer (e.g., Intel Core i5 CPU, 16.0 GB RAM).
Procedure:
- Molecular Modeling: Construct a 3D model of the Schiff base ligand using the building tools in your computational software.
- Geometry Optimization: Perform an initial geometry optimization using a molecular mechanics force field (e.g., MM+). Use this optimized structure as the starting point for a more precise optimization using the AM1 semi-empirical method. Set the convergence limit for the root-mean-square (RMS) gradient to approximately 0.05 kcal/mol/Ångstrom.
- Frequency Calculation: On the fully optimized geometry, perform a vibrational frequency calculation using the AM1 Hamiltonian. This will generate the theoretical IR spectrum.
- Spectral Analysis: Compare the simulated spectrum with the experimental FT-IR spectrum of your synthesized compound. Focus on key regions: the imine (C=N) stretch (~1600-1650 cm⁻¹), aromatic C=C stretches (1400–2000 cm⁻¹), and other characteristic vibrations of functional groups present in your molecule [3]. The theoretical frequencies may require a scaling factor for direct comparison.
Protocol 3: DNA Binding and Cleavage Studies
Aim: To evaluate the interaction of the synthesized Cu(II)-Schiff base complex with DNA and assess its DNA cleavage efficacy [43].
Materials:
- Reagents: Calf Thymus DNA (ct-DNA), pBR322 plasmid DNA, Tris-HCl buffer, ethidium bromide, metal complex solution.
- Equipment: UV-Vis spectrophotometer, fluorescence spectrophotometer, circular dichroism (CD) spectropolarimeter, gel electrophoresis apparatus.
Procedure:
- DNA Binding (Spectroscopic Titration): Prepare a solution of ct-DNA in Tris-HCl buffer (pH 7.4). Record the UV-Vis spectrum of the Cu(II) complex. Titrate the complex solution with aliquots of the DNA solution. Observe changes in the absorption bands (hypochromism or hyperchromism) and shifts in wavelength to determine the binding strength and mode.
- DNA Binding (Fluorescence Quenching): Prepare a DNA solution saturated with a fluorescent intercalator like ethidium bromide (EtBr). Titrate this EtBr-DNA system with the Cu(II) complex and monitor the fluorescence quenching, which indicates the complex is displacing EtBr from the DNA base pairs.
- DNA Cleavage (Gel Electrophoresis): Incubate the pBR322 plasmid DNA with varying concentrations of the Cu(II) complex in an appropriate buffer. Include controls with DNA alone and with a known cleavage agent. After incubation, run the samples on an agarose gel. Stain with EtBr and visualize under UV light. A shift from the supercoiled (Form I) to the nicked open-circular (Form II) or linear (Form III) form indicates DNA cleavage.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Schiff Base Complex Studies
| Reagent / Material | Function and Explanation |
|---|---|
| Salicylaldehyde Derivatives | A common carbonyl precursor for Schiff base synthesis; the ortho-hydroxy group enables chelation with metal ions, forming stable 5-membered rings [41]. |
| Aromatic Diamines (e.g., 1,2-phenylenediamine) | Primary amine components that form bifunctional Schiff base ligands capable of bridging metal centers, creating complexes with varied geometries [42]. |
| Copper(II) Acetate / Chloride | Common sources of Cu(II) ions; known to form complexes with significant biological activity, including DNA binding and cleavage, and cytotoxicity [43]. |
| Calf Thymus (ct-DNA) | A standard model substrate for in vitro DNA binding studies using techniques like UV-Vis, fluorescence, and CD spectroscopy to determine binding constants and mode of interaction [43]. |
| pBR322 Plasmid DNA | A circular DNA vector used to study the DNA cleavage or damage activity of metal complexes via gel electrophoresis, visualizing the conversion from supercoiled to nicked or linear forms [43]. |
| Tris-HCl Buffer | A standard biochemical buffer (typically at pH 7.4) used to maintain physiological conditions during DNA binding and cleavage experiments [43]. |
Infrared (IR) spectroscopy is an indispensable tool for identifying functional groups in complex materials, from small organic molecules to novel two-dimensional carbon materials [3]. The technique is highly sensitive to specific functional groups, as different bonds absorb infrared radiation at characteristic frequencies [3]. This application note provides detailed protocols and analytical frameworks for interpreting C=C stretching vibrations, O-H bands, and hydrogen bonding effects within the context of semi-empirical methods for IR spectra simulation. We focus particularly on challenges associated with functional group analysis in 2D materials and the evolving role of computational approaches in spectral interpretation.
Theoretical Background
Characteristic IR Vibrational Frequencies
The identification of functional groups relies on recognizing characteristic absorption bands in specific regions of the IR spectrum. The table below summarizes the key vibrational frequencies relevant to this application note.
Table 1: Characteristic IR Absorption Frequencies of Key Functional Groups
| Functional Group | Vibration Mode | Frequency Range (cm⁻¹) | Band Characteristics |
|---|---|---|---|
| Alcohol O-H | O-H Stretch | 3200-3600 | Very intense, broad band [44] [45] |
| Carboxylic Acid O-H | O-H Stretch | 3500-2500 | Very broad, often dominates spectrum [46] |
| Free O-H | O-H Stretch | 3700-3590 | Sharp small absorption peaks [47] |
| Alkene | C=C Stretch | 1680-1640 | Varies with substitution pattern [45] |
| Aromatic | C=C Stretch (in-ring) | 1600-1585 and 1500-1400 | Multiple bands due to ring vibrations [45] |
| Terminal Alkyne | C≡C Stretch | 2100-2260 | Strong intensity, position influenced by substituents [48] |
| Alcohol | C-O Stretch | 1260-1050 | Strong intensity [44] [45] |
Hydrogen Bonding Effects
Hydrogen bonding significantly influences IR spectra, particularly for O-H and N-H groups. When O-H groups form hydrogen bonds, the stretching vibration frequency moves toward lower wavenumbers, with the magnitude of the shift corresponding to hydrogen bond strength [49] [47]. This phenomenon results in broad absorption bands because hydrogen bonding between hydroxyl groups leads to variations in O-H bond strength [46]. For coal samples, the presence of numerous hydrogen bonds creates a broadened absorption peak near 3400 cm⁻¹, while free O-H groups without hydrogen bonding appear as sharp small absorption peaks between 3700 and 3590 cm⁻¹ [47].
Experimental Protocols
Sample Preparation Methods
Proper sample preparation is critical for obtaining accurate IR spectra, particularly when analyzing hygroscopic samples or investigating hydrogen bonding.
Table 2: Key Research Reagent Solutions and Materials
| Reagent/Material | Function/Application | Critical Processing Notes |
|---|---|---|
| Potassium Bromide (KBr) | Diluent for pellet press method; transparent to IR radiation | Must be dried at 120°C for 24h and stored in desiccator; spectral grade recommended [47] |
| Fluorolube | Paste medium for mull technique; alternative to KBr | Reagent grade; used without further purification; avoids moisture interference [47] |
| Chloroform | Organic solvent for solution spectroscopy | Must be rigorously dried; remove alcohol added as antioxidant [49] |
| Deuterated Solvents | Solvent for verification of assignments through deuterium substitution [49] | Follow standard safety and handling protocols |
KBr Pellet Press Method with Drying Protocol
- Preparation: Dry spectral grade KBr powder at 120°C for 24 hours and store in a desiccator for standby use [47].
- Weighing: Precisely weigh approximately 1 mg of dried coal sample and 200 mg of dried KBr [47].
- Mixing: Grind and mix the materials thoroughly to ensure homogeneous distribution.
- Pellet Formation: Compress the mixture under high pressure to form a transparent pellet.
- Drying Considerations: For hydroxyl group analysis, employ additional vacuum drying during pellet preparation to remove water absorbed by KBr [47].
- Spectral Acquisition: Immediately analyze the pellet to minimize moisture absorption.
Paste Method (Fluorolube Mull Technique)
- Sample Preparation: Crush the sample to pass through a 100-mesh sieve and dry at 105°C according to national standards [47].
- Mull Formation: Mix the dried sample with Fluorolube to create a uniform paste.
- Application: Sandwich the paste between IR-transparent windows.
- Advantage: This method avoids moisture interference from KBr, providing more accurate O-H analysis [47].
In Situ Infrared Spectroscopy
- Atmosphere Control: Place sample in a nitrogen atmosphere to eliminate atmospheric moisture interference [47].
- Thermal Treatment: Heat to 100°C at 5°C/min and maintain for 1 hour to remove moisture [47].
- Spectral Recording: Collect diffuse reflectance infrared Fourier transform (DRIFT) spectra online during thermal treatment [47].
Data Collection Parameters
- Spectral Range: Collect data from 4000-500 cm⁻¹ to cover the characteristic functional group region [50].
- Resolution: Set to 4 cm⁻¹ for most applications to balance detail and signal-to-noise ratio.
- Scan Accumulation: Minimum of 32 scans to improve signal-to-noise ratio.
- Background Subtraction: Collect background spectrum under identical conditions without sample.
- Temperature Control: Rigorously control cell temperature as hydrogen bonding is temperature-sensitive [49].
Computational Approaches
Semi-Empirical Methods for IR Simulation
Semi-empirical methods provide a balanced approach for calculating IR spectra, offering reasonable accuracy with reduced computational cost compared to ab initio methods.
Diagram 1: Computational workflow for semi-empirical IR simulation
Geometry Optimization with AM1 Method
- Initial Structure Generation: Create initial molecular structure using standard bond lengths (e.g., C-C distance of 1.397 Å, C-H distance of 1.084 Å, bond angles of 120° for PAHs) [3].
- Preliminary Optimization: Perform initial geometry optimization using molecular mechanics.
- Semi-Empirical Optimization: Employ the AM1 semi-empirical method for refined optimization [3].
- Convergence Criteria: Set gradient limit for energy change to 0.05 kcal/mol/Ångstrom as convergence criterion [3].
- Stable Structure Confirmation: Verify optimization completion through energy stabilization in output file.
Vibrational Frequency Calculation
- Force Calculation: Perform force calculation on optimized geometry to obtain vibrational modes [3].
- Frequency Comparison: Compare calculated frequencies with experimental data.
- Error Assessment: If match between calculated and experimental frequencies exceeds 10%, consider semi-empirical method unsuitable for that vibrational mode [3].
- Intensity Calculation: Calculate IR intensities and spectral intensity patterns.
- Ground State Calculation: Perform single-point calculation using ZINDO method to obtain molecular orbital coefficients and eigenvalues [3].
- Configuration Interaction: Execute configuration interaction calculation to determine transition energies [3].
- Spectral Generation: Generate electronic transition spectrum with wavelengths and oscillator strengths [3].
Emerging Machine Learning Approaches
Recent advances introduce machine learning models like AIQM2 that provide IR spectra with accuracy approaching density functional theory (DFT) but with speeds comparable to semi-empirical GFN2-xTB method [25]. These approaches use harmonic approximation with empirically determined scaling factors and offer visualization tools for interpreting IR spectra by vibrational normal modes [25].
Data Interpretation Guidelines
Analyzing O-H Stretching Regions
The O-H stretching region provides critical information about hydrogen bonding and molecular environment.
Free vs. Hydrogen-Bonded O-H:
Differentiating Alcohols and Carboxylic Acids:
Interference Identification:
Interpreting C=C and C≡C Stretching Regions
C=C Stretching Vibrations:
C≡C Stretching Vibrations:
Strategic Spectrum Analysis
Rather than attempting to assign every peak, focus analysis on key regions that provide the most diagnostic information:
High Priority Regions:
Secondary Diagnostic Regions:
Contextual Considerations:
- Consult molecular formula to determine possible functional groups
- Calculate degrees of unsaturation to guide expectations [46]
- Consider sample history and preparation methods
Application to 2D Materials
Functional group analysis in 2D materials like graphene quantum dots (GQDs) presents unique challenges as IR spectra typically assume 3D materials [3]. Semi-empirical methods enable investigation of functionalization at edges and center surfaces of 2D carbon materials [3]. For GQDs, IR analysis focuses on:
- 1400-2000 cm⁻¹ region for C=C stretching mode
- 3000-3500 cm⁻¹ for C-H and O-H stretching [3]
- Functional groups at edges of GQDs significantly alter energy levels, particularly HOMO-LUMO orbitals [3]
This application note provides comprehensive protocols for functional group analysis focusing on C=C stretching, O-H bands, and hydrogen bonding effects. Proper sample preparation, particularly rigorous drying for O-H analysis, is essential for accurate interpretation. Semi-empirical computational methods offer valuable support for spectral interpretation, especially for complex materials like 2D carbon systems. The integration of experimental precision with computational prediction creates a powerful framework for functional group identification across diverse molecular systems.
Tautomer and Protonation State Determination in Drug-like Molecules
In computer-aided drug discovery, accurately predicting the dominant tautomeric and protonation states of small molecules is a critical and persistent challenge. A tautomer is defined as a constitutional isomer that can spontaneously convert to another via the movement of a proton and rearrangement of bonds, a process known as tautomerization [52]. It is estimated that more than a quarter of all marketed drugs can exhibit tautomerism, with analyses suggesting that 10-30% of potential drug-like molecules have viable tautomeric forms [52]. The protonation state refers to the specific sites on a molecule that have gained or lost protons under given pH conditions.
The biological activity of a drug candidate can be dramatically altered by tautomerism, as this process can convert a hydrogen bond donor into an acceptor, or vice versa, fundamentally changing its interaction with a protein target [52] [53]. Despite its importance, the prediction of these states remains difficult due to the scarcity of extensive experimental databases and the computational expense of high-level quantum mechanical (QM) calculations [52] [54]. This application note details modern computational protocols, with a specific focus on the role of semi-empirical methods and infrared (IR) spectra simulation, to address this challenge within a drug discovery workflow.
Key Concepts and Challenges
The Biological Significance of Tautomerism
Tautomerism is not merely a chemical curiosity; it has direct implications for biological activity. In nucleic acids, tautomerization of base pairs can lead to non-Watson-Crick pairing, resulting in spontaneous mutagenesis and genetic instability [52]. For small molecule drugs, the bioactive tautomer bound to a protein target may not be the most stable form in aqueous solution, making its prediction essential for rational drug design [53]. The keto-enol tautomerism of acetone provides a simple example, where the keto form is more stable due to differences in bond dissociation energies [52]. However, for complex drug-like molecules, factors such as electrostatic effects, steric hindrance, and intramolecular hydrogen bonding must be carefully considered [52].
Computational Challenges
The primary challenge in tautomer prediction lies in the accurate calculation of the small free energy differences between tautomeric forms, which often fall within 1-2 kcal/mol [54]. These small differences can be easily compensated by interactions with protein targets, making it crucial to consider multiple potential states. State-of-the-art algorithms generally involve two steps: (1) enumeration of all possible tautomers, and (2) prediction of the dominant form or estimation of tautomer populations [52]. Empirical methods, which often rely on Hammett-Taft linear free energy relationships, can suffer from limited accuracy due to under-coverage of chemical space in parameterization databases [52] [54]. While quantum mechanics-based approaches offer improved accuracy, their computational cost has traditionally rendered them impractical for large-scale virtual screening [52].
Computational Approaches and Methodologies
A range of computational methods are available for tautomer and protonation state prediction, each with its own trade-offs between accuracy, speed, and generality. The table below summarizes the key characteristics of these approaches.
Table 1: Comparison of Computational Methods for Tautomer and Protonation State Prediction
| Method Category | Examples | Typical Application | Relative Speed | Key Considerations |
|---|---|---|---|---|
| Empirical/Rule-based | Epik Classic [52] [54], Protoss [55] | High-throughput pre-screening, database curation | Very Fast | Limited by database coverage; may lack accuracy for novel scaffolds [52]. |
| Semi-empirical QM | AM1 [3], PM6, PM7, GFN2-xTB [56] | Intermediate accuracy screening, IR spectra simulation [3] | Fast | Good balance of speed and accuracy; can be enhanced with machine learning [3] [25]. |
| Density Functional Theory (DFT) | M06-2X [54], ωB97X [56] | High-accuracy benchmarking, final validation | Slow | High computational cost but considered a gold standard for energy calculations [54]. |
| Hybrid QM/ML | AIQM1 [56] [25], AIQM2 [25], QDπ [56] | High-accuracy screening and prediction | Medium | Aims for DFT-level accuracy at near-semi-empirical cost; highly promising [56] [25]. |
| Quantum Computing | VQE with qubit-efficient encoding [52] | Proof-of-concept for specific systems | Very Slow (on current hardware) | Currently limited by hardware but may offer long-term potential [52]. |
Special Focus: Semi-Empirical Methods for IR Spectra Simulation
Infrared (IR) spectroscopy is a powerful technique for identifying functional groups and, by extension, inferring tautomeric states. However, interpreting IR spectra for 2D materials or complex molecules can be challenging [3]. Semi-empirical quantum mechanical methods, such as the Austin Model 1 (AM1), provide a viable pathway for simulating IR spectra to assist in this analysis.
The simulation of IR spectra for pyrene-like molecules, which serve as models for graphene quantum dots (GQDs), demonstrates the utility of this approach. The AM1 method can successfully predict infrared intensities and spectral patterns, particularly in the aromatic C=C stretching region (1400–2000 cm⁻¹) and the C–H/O–H stretching region (3000–3500 cm⁻¹) [3]. This allows researchers to correlate spectral features with specific functionalizations at the edge or center of the molecular surface. Furthermore, combining semi-empirical geometry optimization and frequency calculations with ZINDO method calculations for electronic transitions provides a comprehensive picture of the molecule's properties, helping to confirm its identity as a quantum dot material [3].
Recent advancements have integrated machine learning with semi-empirical methods to create next-generation tools. The AIQM models, for instance, are hybrid quantum mechanical/machine learning potentials that target coupled-cluster level accuracy at a computational speed close to semi-empirical methods like GFN2-xTB [25] [24]. These models can simulate molecular IR spectra with accuracy close to DFT but at a significantly reduced computational cost, making them highly suitable for drug discovery applications [25].
Detailed Experimental Protocols
Protocol 1: Tautomer State Prediction with Semi-Empirical Methods and IR Simulation
This protocol outlines the steps for using semi-empirical methods to predict the stable tautomeric state of a drug-like molecule, with verification via IR spectra simulation.
Table 2: Key Research Reagent Solutions
| Item Name | Function/Description | Example Software/Package |
|---|---|---|
| Molecular Builder | Creates and edits initial 3D molecular structures. | Avogadro, ChemDraw 3D |
| Semi-empirical QM Code | Performs geometry optimization, frequency, and energy calculations. | MOPAC, Winmostar, ORCA |
| Hybrid QM/ML Platform | Provides high-accuracy spectra and energies. | MLatom [25] [24] |
| Tautomer Enumeration Tool | Generates all possible tautomeric and protonation states. | Epik, Protoss, KNIME |
Procedure:
- Initial Structure Preparation: Construct a reasonable 3D model of the input molecule using a molecular builder. Ensure proper bond orders and atom types.
- Tautomer and Protonation State Enumeration: Use a tool like Protoss to generate all possible tautomers and protonation states relevant to the physiological pH range. Protoss uses a consistent chemical model to handle these states for both proteins and ligands, considering the optimal hydrogen bonding network [55].
- Geometry Optimization: For each enumerated structure, perform a geometry optimization using a semi-empirical method (e.g., AM1 or PM6). The convergence limit for the gradient is typically set to 0.05 kcal/mol/Ångstrom [3].
- Frequency Calculation: Upon obtaining a stable optimized structure, conduct a vibrational frequency calculation using the same semi-empirical method. This calculation will confirm the structure is at a local energy minimum (no imaginary frequencies) and provide the necessary data for IR spectrum simulation.
- Single Point Energy Calculation: Perform a more accurate single-point energy calculation on the optimized geometry. For higher accuracy, use a hybrid QM/ML method like AIQM1 or a DFT functional. The relative energies of the different tautomers will determine their predicted stability and population.
- IR Spectrum Analysis: Analyze the simulated IR spectra of the low-energy tautomers. Focus on key spectral regions that are characteristic of specific functional groups (e.g., C=O stretch vs. O-H stretch in keto-enol tautomers) to provide spectroscopic evidence for the predicted structures [3].
- Validation (Optional but Recommended): Where possible, compare the simulated IR spectra of the most stable tautomer(s) with experimental data to validate the computational predictions.
The following workflow diagram illustrates this protocol:
Workflow for Tautomer State Prediction
Protocol 2: Structure-Based Protonation State Prediction for Protein-Ligand Complexes
This protocol is used when the 3D structure of the protein-ligand complex is available, aiming to predict the most likely protonation states of the ligand and the protein's binding site residues.
Procedure:
- Protein and Ligand Preparation: Obtain the 3D structure of the protein-ligand complex from a source like the Protein Data Bank (PDB). Use a tool like Protoss to assign bond orders, atom types, and infer missing heavy atoms for non-standard residues and the ligand [55].
- Initial Hydrogen Placement: Generate initial hydrogen atom coordinates based on idealized geometries from the atom types. At this stage, hydrogens are placed in neutral, normalized states for all ionizable and tautomeric groups [55].
- Enumeration of Alternative States: Identify all substructures with variable hydrogen positions. This includes:
- Rotatable hydrogens (e.g., on hydroxyl, thiol, or acyclic amine groups).
- Alternative tautomeric forms of the ligand and protein residues (e.g., His).
- Different protonation states of acidic and basic groups.
- Alternative orientations of water molecules within the binding site [55].
- Scoring and Optimization: Evaluate all possible combinations of these alternative states. An empirical scoring function is used to identify the configuration that optimizes the overall hydrogen bonding network, considering both the quality of hydrogen bonds and the intrinsic stability of the different chemical groups [55].
- Output Optimal States: The algorithm outputs the protein-ligand complex with hydrogen coordinates assigned according to the most probable protonation and tautomeric states, ready for further computational studies like molecular docking or binding free energy calculations.
Data Interpretation and Analysis
When analyzing results, the relative potential energy difference between tautomers is the primary factor determining the equilibrium direction [52]. However, for drug discovery, it is often critical to consider not only the global minimum but also low-energy tautomers (e.g., within ~2 kcal/mol), as these may be populated at room temperature and could be the bioactive form.
In the analysis of simulated IR spectra, focus on diagnostic regions. For instance, a strong absorption band around 1700 cm⁻¹ is typical of a carbonyl (C=O) stretch in a keto tautomer, while a broad band in the 3000-3500 cm⁻¹ region can indicate an O-H stretch from an enol or carboxylic acid group [3]. Significant shifts in the aromatic C=C stretching region (1400–2000 cm⁻¹) upon functionalization can also provide insights into the molecular structure [3].
Table 3: Troubleshooting Guide for Computational Prediction
| Problem | Potential Cause | Solution |
|---|---|---|
| Implausible low-energy tautomer | Inadequate conformational sampling or trapped in local minimum. | Verify optimization converged. Perform a conformational search before tautomer optimization. |
| Mismatch between predicted and experimental IR | Harmonic approximation errors, anharmonicity, or solvent effects neglected. | Use methods that account for anharmonicity (e.g., molecular dynamics) [57] or apply empirical scaling factors. |
| High computational cost for large molecules | Using high-level DFT on a large, flexible molecule. | Switch to a faster semi-empirical (GFN2-xTB) or hybrid QM/ML (AIQM) method [56] [25]. |
The reliable prediction of tautomeric and protonation states is a non-trivial yet essential component of modern drug discovery. While empirical methods offer speed, and pure QM methods provide high accuracy, semi-empirical methods strike a practical balance for many applications. The integration of machine learning, as seen in hybrid models like AIQM and foundation models like MACE4IR, is pushing the boundaries of what is possible, enabling faster and more accurate simulations of key experimental data such as IR spectra [25] [57]. By adopting the protocols outlined in this document, researchers can make more informed decisions in virtual screening and structure-based design, ultimately increasing the efficiency of the drug discovery pipeline.
Infrared (IR) spectroscopy serves as a powerful experimental technique for molecular structure identification and characterization of chemical properties. In computational chemistry, several theoretical approaches have been developed to simulate and predict IR spectra, each offering different balances between computational cost and accuracy. Traditional quantum chemical calculations face significant challenges, with density functional theory (DFT) providing good accuracy at high computational cost, while semi-empirical methods offer greater speed but often with insufficient accuracy for many applications [24]. This application note examines three software platforms—HyperChem, MOPAC, and MLatom—that implement different computational approaches for IR spectra simulation, with particular relevance to researchers investigating semi-empirical methods.
The recent integration of machine learning (ML) with computational chemistry has created new opportunities for overcoming the limitations of traditional approaches. ML-enhanced methods, particularly the AIQM series, now offer pathways to achieve accuracy approaching coupled-cluster levels while maintaining speeds comparable to semi-empirical methods [58]. These developments are particularly valuable for researchers in drug development who require both rapid screening capabilities and high predictive accuracy for molecular behavior.
Platform Specifications and Capabilities
Table 1: Platform overview and key characteristics
| Platform | Computational Method | Target Accuracy | Computational Speed | IR Simulation Approach |
|---|---|---|---|---|
| MLatom | AIQM (ML-enhanced quantum mechanics) | CCSD(T)/CBS level accuracy [59] | Close to GFN2-xTB semi-empirical method [24] | Harmonic approximation with scaled frequencies; anharmonic via MD and VPT2 [24] |
| HyperChem | Semi-empirical, Ab Initio, DFT | Varies with method selection | Varies with method selection | Multiple quantum chemistry methods |
| MOPAC | Semi-empirical | Semi-empirical accuracy | Fast calculations | Semi-empirical quantum chemistry |
Table 2: Performance benchmarks for IR spectra simulation
| Platform/Method | Accuracy Relative to Experiment | Computational Cost | Key Strengths |
|---|---|---|---|
| MLatom (AIQM) | Close to DFT accuracy [58] | Near semi-empirical speed [58] | Optimal balance of speed and accuracy |
| DFT | High (reference) | High computational cost [24] | High accuracy for diverse systems |
| Semi-empirical | Lower accuracy [24] | Orders of magnitude faster than DFT [24] | Rapid screening of large molecular sets |
Hardware/Software Requirements
Table 3: Platform requirements and availability
| Platform | System Requirements | License Model | Interface Options |
|---|---|---|---|
| MLatom | Web browser access or local installation [24] | Open source | Web interface, command line [60] |
| HyperChem | Windows 98 to Windows 7 [61] | Commercial (10-day evaluation available) [61] | Graphical user interface |
| MOPAC | Not specified in sources | Commercial | Interface via HyperChem [62] |
MLatom Implementation and Protocols
AIQM Model Theory and Implementation
MLatom implements artificial intelligence-enhanced quantum mechanical methods, specifically the AIQM series, which target coupled-cluster quality accuracy [63]. These models are designed to overcome the traditional accuracy-speed tradeoff by leveraging machine learning to approximate high-level quantum mechanical calculations. The AIQM approach provides "accuracy close to DFT and the speed close to a semi-empirical GFN2-xTB method" [58], making it particularly valuable for IR spectra simulation in research applications where both efficiency and predictive reliability are essential.
For IR spectra simulation, MLatom employs a harmonic oscillator approximation with frequency scaling factors fitted to experimental data [59]. This approach maintains computational efficiency while ensuring results that closely align with experimental observations. The platform also supports more advanced anharmonic spectra simulations through molecular dynamics and vibrational perturbation theory (VPT2) implementations [24], offering researchers flexibility in selecting the appropriate level of theory for their specific application needs.
Step-by-Step Protocol for IR Spectra Simulation
Protocol 1: MLatom IR Spectrum Calculation via Web Interface
Input Preparation: Prepare a molecular structure file in XYZ format or use the built-in molecular builder if available.
Method Selection:
- Select the AIQM model for optimal balance of accuracy and speed
- Alternatively, choose DFT methods for higher accuracy at greater computational cost
- For large systems, consider semi-empirical methods available through the platform
Calculation Execution:
- Upload input file to MLatom@XACS web interface [60]
- Submit calculation job
- Monitor job status until completion
Output Analysis:
Validation:
- Compare with experimental data if available
- Apply statistical metrics to assess prediction quality
Protocol 2: Advanced Anharmonic Calculation
- Setup: Access anharmonic options in MLatom implementation
- Method Selection: Choose between molecular dynamics or VPT2 approaches
- Parameters: Set appropriate simulation parameters (temperature, time step)
- Execution: Run extended calculation
- Analysis: Compare harmonic and anharmonic results to assess improvements
HyperChem and MOPAC Implementations
HyperChem Setup and Configuration
HyperChem provides a sophisticated molecular modeling environment that unites 3D visualization with quantum chemical calculations, molecular mechanics, and dynamics [64]. For researchers focusing on semi-empirical methods for IR spectra simulation, HyperChem offers multiple computational approaches within an integrated graphical interface.
Installation and Licensing Protocol:
- Download: Obtain the HyperChem 8.0.10 evaluation copy from the official provider [61]
- Installation: Run the self-extracting executable and install following the setup wizard
- Serial Number: Input the serial number 12-800-1501799999 when prompted [61]
- Activation:
- Execute HyperChem to invoke the "Client Activator"
- Select "Activate Now" to generate a locking code
- Email the locking code and serial number to [email protected] to request a temporary evaluation license [61]
- License Installation:
- Receive the license file (*.lic) via email
- Place the file in an accessible location on your computer
- Execute HyperChem again and navigate to the license file when prompted
- Complete activation for 10-day evaluation usage [61]
MOPAC Integration and Semi-Empirical Methods
HyperChem includes interfaces to third-party applications including MOPAC, with specific compatibility with Mopac2009 [62]. This integration enables researchers to leverage MOPAC's semi-empirical methods directly within the HyperChem environment, combining the user-friendly graphical interface with MOPAC's computational efficiency.
Protocol for MOPAC Calculation via HyperChem:
- Structure Preparation: Build or import molecular structure using HyperChem's visualization tools
- Method Selection:
- Access the computational method menu
- Select MOPAC from the semi-empirical options
- Choose appropriate parameter set (AM1, PM3, etc.)
- Calculation Setup:
- Specify calculation type (vibrational frequencies)
- Set convergence criteria and other parameters
- Job Execution:
- Run calculation locally or distribute to computational servers
- HyperChem can farm out computations to Linux servers [62]
- Results Analysis:
- View vibrational frequencies and intensities in output
- Visualize normal modes using molecular animation tools
- Export data for further analysis
Research Reagent Solutions
Table 4: Essential computational reagents for IR spectra simulation
| Research Reagent | Function/Purpose | Implementation Examples |
|---|---|---|
| AIQM Models | ML-enhanced quantum mechanical method targeting CCSD(T) accuracy [58] | MLatom's AIQM1 and AIQM2 for balanced accuracy/speed |
| Scaling Factors | Empirical correction for harmonic approximation inaccuracies | Frequency scaling in MLatom fitted to experimental data [59] |
| GFN2-xTB | Fast semi-empirical method for large systems | Reference method for speed comparison in MLatom [24] |
| VPT2 Implementation | Anharmonic correction for improved accuracy | Advanced spectral simulation in MLatom [24] |
| Normal Mode Visualization | Interpretation and assignment of IR bands | MLatom tool for visualizing vibrations [58] |
Workflow Integration and Decision Pathways
The evolving landscape of computational tools for IR spectra simulation offers researchers multiple pathways for investigating molecular properties. HyperChem provides a comprehensive graphical environment with support for semi-empirical methods through its MOPAC interface, while MLatom represents the cutting edge of machine-learning enhanced quantum mechanical simulations. For research focused on semi-empirical methods, MLatom's AIQM approach offers a significant advancement, delivering accuracy approaching DFT methods while maintaining computational speeds comparable to traditional semi-empirical approaches [58]. This balance makes it particularly valuable for drug development professionals requiring both rapid screening capabilities and reliable predictive accuracy for molecular behavior and spectroscopic properties.
The protocols and implementations detailed in this application note provide researchers with practical guidance for selecting and applying these tools within their IR spectra simulation workflows. As machine learning continues to transform computational chemistry, platforms like MLatom are poised to become increasingly central to research investigating the relationship between molecular structure and spectroscopic behavior.
Overcoming Challenges: Accuracy Improvement and Error Mitigation
In the research of semi-empirical methods for IR spectra simulation, systematic errors arise from two primary sources: inaccuracies in the potential energy surface and the neglect of anharmonic effects [65]. Addressing these errors is critical for achieving accurate, experimentally comparable results. This document outlines core methodologies, including frequency scaling and parameter optimization, to correct these systematic deviations, providing structured protocols for researchers and scientists in drug development.
Core Concepts and Quantitative Data
Frequency Scaling Fundamentals
The calculated harmonic frequencies (( \omega{calc} )) from quantum mechanical methods are typically higher than observed fundamental frequencies (( \nu{obs} )) due to the noted systematic errors. A standard correction is the application of a multiplicative scaling factor, ( s ) [65]:
[ \nu{calc} = s \omega{calc} ]
Mathematically, this scaling is equivalent to scaling the potential energy surface by ( s^2 ) [65]. For more advanced treatments, particularly with high-quality calculations, non-linear mass scaling proves more effective than simple frequency scaling, especially for predicting vibrational fundamentals and zero-point energies of isotopologues [65].
Performance of Parameterized Methods
Comprehensive parameter optimization, as demonstrated in the development of the PM6 method, significantly reduces errors across a wide range of elements and compounds. The table below summarizes the performance of various semi-empirical and other computational methods for predicting heats of formation, demonstrating the impact of rigorous parameterization.
Table 1: Average Unsigned Error (AUE) for Heats of Formation Prediction (kcal mol⁻¹)
| Method | AUE for 1,373 Compounds (H, C, N, O, F, P, S, Cl, Br) | AUE for 4,492 Species |
|---|---|---|
| PM6 | 4.4 | 8.0 |
| RM1 | 5.0 | - |
| B3LYP/6-31G* | 5.2 | - |
| PM5 | 5.7 | - |
| PM3 | 6.3 | - |
| HF/6-31G* | 7.4 | - |
| AM1 | 10.0 | - |
Data sourced from [66]. The subset of 1,373 compounds involves only the elements H, C, N, O, F, P, S, Cl, and Br.
The optimization process for PM6 used a considerably larger training set (over 9,000 separate species) compared to its predecessor PM3 (approximately 800 species), contributing to its improved accuracy [66]. Long-standing faults in AM1 and PM3 were corrected, and significant improvements were made in geometry prediction [66].
Experimental Protocols
Protocol A: Applying Frequency Scaling Factors
This protocol details the steps to derive and apply a frequency scaling factor for a set of diatomic molecules.
Table 2: Key Research Reagents for Frequency Scaling
| Item Name | Function/Description |
|---|---|
| Reference Data Set | A collection of diatomic molecules with experimentally known fundamental frequencies (( \nu{obs} )) and calculated harmonic frequencies (( \omega{calc} )). |
| Computational Model | The specific ab initio or density functional theory (DFT) method and basis set used to calculate ( \omega_{calc} ). |
| Error Minimization Algorithm | A routine (e.g., least squares fitting) to find the parameter ( s ) that minimizes the difference between ( \nu{calc} ) and ( \nu{obs} ). |
Procedure:
- Select Molecules and Calculate: Choose a representative set of diatomic molecules. For each molecule, perform a frequency calculation using your chosen computational model to obtain the harmonic frequency, ( \omega_{calc} ) [65].
- Gather Experimental Data: Compile the experimentally observed fundamental frequencies, ( \nu_{obs} ), for the same set of molecules from reliable spectroscopic sources [65].
- Optimize Scaling Factor: Determine the optimal scaling factor ( s ) by minimizing the sum of squared differences between the calculated and observed frequencies. The optimum scaling factor is given by: [ s = \frac{\sum \omega{calc} \nu{obs}}{\sum \omega{calc}^2} ] The minimized sum of squares is ( y{min} = \sum \nu{obs}^2 - s^2 \sum \omega{calc}^2 ) [65].
- Apply and Validate: Apply the scaling factor ( s ) to new calculations using the same computational model. Validate the accuracy by comparing scaled frequencies against a separate test set of molecules not included in the parameterization.
Protocol B: Global Parameter Optimization for Semi-Empirical Methods
This protocol outlines a generalized workflow for optimizing parameters in semi-empirical methods like PM6, based on reproducing a wide array of reference data.
Table 3: Key Research Reagents for Parameter Optimization
| Item Name | Function/Description |
|---|---|
| Reference Data Compilation | A large, diverse set of experimental and high-level ab initio data, including heats of formation, molecular geometries, dipole moments, and ionization potentials. |
| Core-Core Interaction Modifications | Theoretical improvements to the NDDO Hamiltonian, such as added Gaussian functions, to better model phenomena like hydrogen bonding. |
| Weighting Rules | A system to assign importance (weights) to different types of reference data to ensure balanced optimization across properties. |
Procedure:
- Compile Reference Data: Assemble a large training set of reference data. This should include thermochemical data (e.g., heats of formation from databases like NIST WebBook), molecular geometries (e.g., from the Cambridge Structural Database), and other target properties [66]. Where experimental data is scarce, use results from robust ab initio or DFT calculations (e.g., B3LYP/6-31G(d)) [66].
- Define the Error Function: Construct an error function, ( S ), that quantifies the difference between calculated and reference values. For heats of formation, this can be a root-mean-square fit: [ S = \sqrt{ \frac{1}{N} \sum{j=1}^{N} \left( \Delta H{f,calc,j} - \Delta H{f,ref,j} \right)^2 } ] where ( \Delta H{f,calc} ) is a function of the total semi-empirical energy, ( E{tot} ), and atom counts: ( \Delta H{f,calc} = \sum ni Ci + Cx + k E{tot} ) [66].
- Implement Weighting Rules: Assign appropriate weights to critical but low-energy phenomena. For example, to accurately reproduce hydrogen bond energies (~5 kcal mol⁻¹) when general AUE for ΔHf is 3-5 kcal mol⁻¹, the weighting for the water dimer and monomer must be strategically increased [66].
- Iterative Optimization: Systematically adjust the method's parameters to minimize the global error function, ( S ), across the entire training set. This process may involve correcting specific long-standing faults in prior parameterizations [66].
Protocol C: The Instantaneous Frequencies of Molecules (IFM) Method
The IFM method is a modern, parameterization-free approach for predicting vibrational observables, such as the frequency fluctuation correlation function (FFCF), from molecular dynamics (MD) simulations [67].
Procedure:
- Run Molecular Dynamics (MD): Perform a classical MD simulation of the system of interest (e.g., N-methylacetamide (NMA) in various solvents) [67].
- Sample Snapshots: Extract multiple snapshots from the MD trajectory that represent the system's configurational space [67].
- Compute Instantaneous Frequencies: For each snapshot, calculate the instantaneous vibrational frequency of the mode of interest (e.g., the amide I mode of NMA) using a low-cost but accurate quantum mechanical method like GFN2-xTB. This step bypasses the need for a pre-parameterized frequency map [67].
- Construct the FFCF: Analyze the time series of instantaneous frequencies to compute the frequency fluctuation correlation function, which provides details on the dynamics and heterogeneity of the chemical environment [67].
Workflow Visualization
The following diagram illustrates the logical relationship and workflow between the core protocols described in this document for addressing systematic errors in IR spectra simulation.
Application in Drug Development
Semi-empirical methods with proper error correction are vital in drug development for simulating the IR spectra of complex molecular systems, such as metal complexes of pharmaceutical compounds.
For instance, PM3 semi-empirical method has been successfully used to simulate the IR spectra of Co, Ni, Cu, and Zn complexes with Schiff base ligands derived from sulfa drugs [68]. The method accurately characterized coordination effects on key vibrational modes like ν(C=N) and ν(C–O), showing close agreement with experimental results [68]. This application demonstrates the utility of well-parameterized semi-empirical methods in confirming the molecular structures of potential drug candidates or their complexes, where experimental analysis alone may be insufficient.
Semi-empirical quantum mechanical (SQM) methods offer a compelling compromise between computational cost and electronic structure detail, making them particularly attractive for simulating infrared (IR) spectra of large molecular systems such as those encountered in drug development. However, a significant challenge impedes their routine application: the inherently poor description of non-covalent interactions, especially hydrogen bonding [69] [70]. These interactions are not merely minor details; they are fundamental forces governing molecular self-assembly, protein-ligand binding, and the structural fidelity of biomolecules [71]. In the context of IR spectroscopy, hydrogen bonds directly influence vibrational frequencies and intensities, meaning that an inaccurate theoretical model can lead to severe misinterpretation of experimental spectra [72]. This application note details the limitations of standard SQM methods and provides structured protocols for applying modern correction schemes to achieve accuracy commensurate with more computationally intensive ab initio methods.
Limitations of Semi-Empirical Methods
The primary failure of SQM methods in describing weak interactions stems from their underlying approximations, primarily the Neglect of Diatomic Differential Overlap (NDDO). These approximations lead to systematic errors in the calculation of interaction energies and equilibrium geometries for molecular complexes.
- Hydrogen Bonding Inaccuracy: Uncorrected SQM methods, such as AM1 and PM6, significantly underestimate hydrogen bond energies and fail to reproduce correct intermolecular distances. This results in a distorted picture of the hydrogen-bonding network, which is critical for understanding the IR spectra of biological systems or functionalized materials like graphene quantum dots (GQDs) [3] [69].
- Inadequate Treatment of Dispersion Forces: Long-range dispersion interactions, which are ubiquitous and critical for molecular stacking and stability, are completely absent from standard SQM Hamiltonians [70].
- Orientation-Dependent Errors: The performance of SQM methods is not uniformly poor but varies dramatically with the relative orientation of the interacting molecules. A correction parameterized for a single, optimal geometry may fail for other configurations, limiting its transferability [70].
Table 1: Quantitative Performance of Uncorrected SQM Methods for Noncovalent Interactions.
| SQM Method | Typical H-bond Energy Error (kcal/mol) | Representative Issue |
|---|---|---|
| AM1 | Large Underestimation | Poor description of interaction energies and geometries [69]. |
| PM6 | Large Underestimation | Overestimation of interaction energies and underestimation of equilibrium distances for hydrocarbons [70]. |
| OM3 | Large Underestimation | Generally poor performance for noncovalent complexes without corrections [69]. |
Correction Schemes and Methodologies
To overcome these limitations, several empirical and theoretical correction schemes have been developed. These are broadly categorized into general-purpose corrections and specialized, orientation-aware approaches.
General Empirical Corrections
The most widely used strategy involves augmenting the SQM energy with analytical potential functions that describe the missing interactions.
- PM6-DH and D3H4X: This family of corrections adds empirical terms for dispersion (D) and hydrogen bonding (H) to the PM6 Hamiltonian. The D3H4X correction, for instance, includes a repulsive term for H...H pairs, a complex angular-dependent function for hydrogen bonds, and a specific term for halogen bonding [70]. This approach brings SQM accuracy closer to that of DFT-D methods for standard benchmark sets like S66 [69] [70].
- PMO Method: The Polarized Molecular Orbital (PMO) method enhances the NDDO Hamiltonian by adding polarization functions on hydrogen atoms and a damped dispersion term, leading to improved descriptions of polarization and noncovalent complexation energies [70].
- OMx and ODMx: These orthogonalization-corrected methods incorporate Grimme's D3 dispersion correction with Becke-Johnson damping, which significantly improves their performance for large, dense systems [70].
The PM6-FGC Approach: Importance of Multiple-Orientation Sampling
A recent and advanced approach highlights a critical shortcoming of previous parameterizations: their reliance on training sets containing only the most favorable molecular orientations. The PM6-FGC (Functional Group Corrections) method underscores that well-balanced corrections require sampling multiple orientations to capture the full panorama of atom-pair interactions [70].
The methodology involves:
- Selection of Representative Molecules: Small molecules (e.g., methane, formic acid, ammonia) are chosen to represent key functional groups.
- Generation of Reference Data: Intermolecular Potential Energy Curves (IPECs) are computed for various orientations of each molecular pair using a high-level ab initio reference method (e.g., CCSD(T)/aug-cc-pVTZ or B3LYP-D3/def2-TZVP).
- Derivation of Analytical Corrections: The differences between the reference and PM6 interaction energies are used to fit a general pairwise correction function. This function includes a cutoff to prevent singularities at short distances [70].
Table 2: Comparison of Key SQM Correction Methods.
| Correction Method | Core Philosophy | Applicable SQM Methods | Key Strengths |
|---|---|---|---|
| D3H4X [70] | Empirical potentials for dispersion, H-bond, and halogen-bond. | PM6, others | Good performance on standard benchmarks (e.g., S66). |
| PMO2 [70] | Hamiltonian modification with polarization functions and dispersion. | Proprietary PMO | Accurate for polarization and complexation energies. |
| OMx/ODMx [70] | Orthogonalization-corrected Hamiltonian with D3 dispersion. | OMx, ODMx | Improved Hamiltonian for larger systems. |
| PM6-FGC [70] | Pairwise corrections fitted to multiple-orientation IPECs. | PM6 (transferable) | Superior, well-balanced description across all orientations. |
Experimental Protocols
Protocol 1: Applying the D3H4X Correction for IR Spectrum Simulation
This protocol is suitable for rapid screening and studies where the system is similar to those in standard benchmark sets.
- System Preparation: Obtain the initial molecular geometry of the isolated molecule or complex using a molecular builder. Perform a preliminary geometry optimization using molecular mechanics.
- Geometry Optimization: Employ a SQM method (e.g., PM6) with the D3H4X correction enabled. The calculation is considered converged when the energy gradient falls below a threshold of 0.05 kcal/mol/Å [3].
- Vibrational Frequency Calculation: On the optimized geometry, perform a force calculation to obtain the vibrational normal modes and their associated IR intensities. The D3H4X correction must remain active during this step to ensure the potential energy surface includes the noncovalent interactions.
- Spectrum Analysis: Compare the calculated harmonic vibrational frequencies to experimental data. A scaling factor may be applied. Focus analysis on key regions sensitive to hydrogen bonding, such as the OH stretching region (3000–3500 cm⁻¹) [3].
Protocol 2: Advanced IR Workflow with the PM6-FGC Correction
Use this protocol for systems where orientation-dependent interactions are critical or when the highest accuracy is required for SQM-based IR simulation.
- Reference Data Generation (If needed for new groups): For each relevant functional group pair in your system, generate IPECs using a high-level method (e.g., CCSD(T)/aug-cc-pVTZ) across multiple orientations [70].
- Parameterization (If needed): Fit the differences between the reference and SQM interaction energies to the PM6-FGC analytical function to obtain new parameters for atom pairs [70].
- System Optimization and IR Calculation: Apply the PM6-FGC parameters during the geometry optimization and subsequent vibrational frequency calculation, following the same convergence criteria as in Protocol 1.
- Electronic Transition Analysis (Optional): To link vibrational features to electronic structure, calculate excitation spectra using the ZINDO method on the optimized geometry [3].
The following workflow diagram illustrates the advanced protocol for obtaining accurate IR spectra using orientation-aware corrections:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Software and Computational Tools for SQM-IR Studies.
| Tool Name | Function / Description | Relevance to Protocol |
|---|---|---|
| MOPAC2016/2022 [70] | Software implementing SQM methods (AM1, PM6) and empirical corrections (D3H4). | Core engine for Geometry Optimization and Frequency Calculation (Protocols 1 & 2). |
| Winmostar [3] | A graphical interface and computational package that includes the AM1 method and ZINDO for excitation spectra. | Used for IR simulation and electronic transition analysis (Protocol 1). |
| ORCA [70] | An ab initio quantum chemistry program with high-performance capabilities. | Used for generating reference IPEC data at the CCSD(T) or DFT-D3 level (Protocol 2). |
| MLatom [33] | A platform for incorporating machine learning models (like AIQM) for quantum simulations. | Alternative for achieving CCSD(T)-level accuracy at SQM speed for IR spectra. |
| S66 Database [70] | A benchmark set of 66 noncovalent interaction energies for method validation. | Critical for validating the performance of any applied correction scheme. |
Within the broader context of developing semi-empirical methods for IR spectra simulation, understanding the specialized parameters and analytical approaches for transition metal complexes is paramount. These complexes, central to catalysis, biological systems, and materials science, exhibit unique electronic and vibrational properties that distinguish them from purely organic molecules. This document outlines the critical parameters governing their behavior and provides detailed protocols for their experimental and computational characterization, with a specific focus on integrating these approaches into a robust framework for simulating infrared spectra. The accurate simulation of these spectra requires a nuanced understanding of both the electronic transitions that influence vibrational modes and the practical aspects of data acquisition and fitting.
Theoretical Foundations: Electronic States and Transitions
The electronic spectrum of a transition metal complex provides profound insights into its geometric and electronic structure. The observed transitions can be categorized into several distinct types, each with characteristic intensities and spectral positions.
Types of Electronic Transitions
The primary electronic transitions observed in transition metal complexes are detailed in the table below.
Table 1: Types of Electronic Transitions in Transition Metal Complexes
| Transition Type | Spectral Region | Extinction Coefficient (ε, M⁻¹cm⁻¹) | Description | Molecular Orbital Character |
|---|---|---|---|---|
| d-d (Ligand Field) | Visible/NIR | < 100 | Weak transitions between metal-centered d-orbitals. Forbidden by Laporte rule, but gain intensity through vibronic coupling. [73] | Metal (d) → Metal (d) |
| Charge Transfer (CT) | UV-Visible | > 1,000 | Intense transitions involving electron movement between metal and ligand orbitals. [73] | |
| * • LMCT* | Ligand to Metal Charge Transfer. Common with π-donor ligands (e.g., O²⁻, Cl⁻). [73] | Ligand → Metal | ||
| * • MLCT* | Metal to Ligand Charge Transfer. Common with π-acceptor ligands (e.g., 2,2'-Bipyridine, CO). [73] | Metal → Ligand | ||
| Intraligand | UV | > 10,000 | Very intense transitions localized on the ligand, e.g., π→π*. [73] | Ligand → Ligand |
These transitions are governed by quantum mechanical selection rules that determine their probability and, consequently, their intensity in the absorption spectrum. [73]
- The Spin Selection Rule (ΔS = 0): This rule forbids transitions between states of different total spin multiplicity. For example, a transition from a singlet state (all electrons paired) to a triplet state (two unpaired electrons) is formally forbidden. However, spin-orbit coupling can relax this rule, particularly in heavier metal ions. [73]
- The Laporte Selection Rule: For molecules with a center of inversion (centrosymmetric), this rule forbids transitions between states of the same parity (e.g., g→g or u→u). Since d-orbitals are gerade (g), pure d-d transitions are Laporte-forbidden in centrosymmetric complexes (e.g., perfect octahedral geometry), contributing to their low intensity. This rule is relaxed in non-centrosymmetric complexes (e.g., tetrahedral) or through vibrational distortions. [73]
Experimental Characterization and Protocols
Synthesis of Amino Acid-Based Transition Metal Complexes
The following protocol, adapted from studies on leucine and isoleucine complexes, provides a general method for synthesizing transition metal complexes with bidentate oxygen/nitrogen-donor ligands. [74]
Protocol 1: Synthesis of M(II)-Amino Acid Complexes (M = Co, Ni, Cu, Zn, Cd, Hg)
- Objective: To synthesize and isolate 1:2 (metal:ligand) complexes of transition metals with amino acids.
Principle: Amino acids like leucine and isoleucine act as bidentate ligands, coordinating to metal ions through their deprotonated carboxylate (-COO⁻) and amino (-NH₂) groups in a five-membered chelate ring, forming stable, often colored, complexes in aqueous solution. [74]
Research Reagent Solutions:
- Essential Materials:
- Metal Salts: Hydrated chlorides or acetates of Co(II), Ni(II), Cu(II), Zn(II), Cd(II), and Hg(II).
- Ligands: L-Leucine and L-Isoleucine.
- Solvent: Deionized water.
- Basifying Agent: Sodium hydroxide (NaOH) or potassium hydroxide (KOH) pellets.
- Precipitation Solvent: Methanol or ethanol.
- Essential Materials:
Procedure:
- Ligand Solution Preparation: Dissolve 2 mmol of the amino acid (e.g., ~262 mg of leucine) in a minimal volume of warm deionized water. Add an equimolar aqueous solution of sodium hydroxide (2 mmol in ~10 mL water) to deprotonate the carboxylic acid group and form the sodium salt of the amino acid.
- Metal Solution Preparation: Dissolve 1 mmol of the desired metal salt in ~5 mL of deionized water.
- Complexation: Slowly add the metal salt solution dropwise to the stirred amino acid solution. A color change or the formation of a precipitate may be observed immediately (e.g., pink for Co, sky blue for Ni, blue for Cu). [74]
- pH Adjustment: Adjust the pH of the reaction mixture to ~7-8 using a dilute NaOH solution to ensure complete deprotonation and complex formation.
- Stirring and Heating: Stir the reaction mixture for 1-2 hours at 40-50°C to complete the reaction.
- Isolation: Concentrate the solution on a rotary evaporator or allow slow evaporation at room temperature. Alternatively, precipitate the complex by adding a excess of a water-miscible solvent like methanol or ethanol.
- Purification: Collect the solid product by vacuum filtration, wash with cold ethanol, and dry in a desiccator over anhydrous silica gel.
- Characterization: The complexes are characterized by Elemental Analysis (CHN), FT-IR, UV-Vis, and Thermogravimetric Analysis (TGA). Magnetic susceptibility measurements can confirm paramagnetic (Co, Ni, Cu) or diamagnetic (Zn, Cd, Hg) behavior. [74]
Infrared Spectral Analysis: A Strategic Workflow
Interpreting IR spectra of transition metal complexes requires a systematic approach to identify key functional groups and metal-ligand vibrations.
Protocol 2: Rapid Analysis of IR Spectra for Transition Metal Complexes
- Objective: To quickly identify the presence of major functional groups and metal-ligand bonding from an IR spectrum.
Principle: Prioritize the analysis of high-information regions to avoid being overwhelmed by the "fingerprint" region (500-1400 cm⁻¹). The most diagnostic regions are the O-H/N-H stretch and the C=O stretch. [46]
Workflow:
- Identify "Tongues" (3200-3600 cm⁻¹): Look for broad, rounded peaks indicating O-H stretches from water, alcohols, or carboxylic acids. Sharper peaks in this region can indicate N-H stretches. The breadth of carboxylic acid O-H peaks is distinctive. [46]
- Identify "Swords" (1630-1800 cm⁻¹): Look for sharp, strong peaks indicating C=O stretches. The exact position helps distinguish between free carbonyls (~1700 cm⁻¹ for aldehydes/ketones) and coordinated carbonyls (e.g., in metal carboxylates, which often show asymmetric stretches at 1600-1650 cm⁻¹). [74] [46]
- Check the 3000 cm⁻¹ "Border": Peaks above ~3000 cm⁻¹ suggest the presence of alkene or aromatic C-H stretches, while those below are typical of alkane C-H. [46]
- Probe for Triple Bonds (2050-2250 cm⁻¹): Look for weak-to-medium sharp peaks indicating C≡C or C≡N stretches. [46]
- Analyze Metal-Ligand Vibrations (< 600 cm⁻¹): The region below 600 cm⁻¹ often contains M-O, M-N, and M-Cl stretches, which are direct evidence of coordination. [74] Shifts in ligand vibrational modes upon coordination (e.g., -COO⁻ asymmetric stretch) are also critical indicators.
The logical flow for this analysis can be visualized as follows:
Figure 1: A strategic workflow for the rapid initial analysis of IR spectra.
Computational Approaches for IR Simulation
Spectrum Simulation and Fitting Protocol
For high-resolution IR spectroscopy, simulating and fitting the spectrum allows for the precise extraction of molecular parameters.
Protocol 3: Simulation and Fitting of a High-Resolution IR Spectrum
- Objective: To extract accurate rotational and vibrational constants from a high-resolution IR spectrum by iterative simulation and fitting.
Principle: A theoretical spectrum is simulated using an initial guess for molecular parameters (rotational constants, band origin). The simulated line positions are then compared to the experimental spectrum, and the parameters are refined via a least-squares fitting procedure until the best possible agreement is achieved. [75]
Procedure (using a program like PGOPHER):
- Load Spectrum and Initialize Simulation: Load the experimental spectrum file. Create a new simulation for an appropriate model (e.g., "Linear Molecule" for the ν₂ bending mode of N₂O). [75]
- Manual Parameter Adjustment: Manually adjust key parameters in the simulation to achieve a rough fit.
- Band Origin (Center): Adjust the
Originparameter for the excited vibrational state to align the simulated band center with the experimental one. - Rotational Constants (B): Adjust the rotational constants for the ground (
B'') and excited (B') states to match the spacing of the P and R branches. The mouse-adjust function can be useful for this. [75] - Band Type: For a bending vibration, set the upper state symmetry to
Πto introduce a Q branch. [75]
- Band Origin (Center): Adjust the
- Line Assignment:
- Right-click on a simulated peak you believe corresponds to an experimental peak.
- In the line list window, drag across the corresponding experimental peak with the right mouse button to measure its position and assign the quantum numbers. [75]
- Repeat for multiple peaks, preferably starting with low-J lines near the band head.
- Test Assignments: Use the "Test" function to plot the differences (residuals) between assigned experimental and calculated line positions. Incorrect assignments will show as large, systematic deviations. [75]
- Least-Squares Fitting:
- In the constants window, set the "Float" option to "yes" for the parameters to be refined (e.g.,
B',B'',Origin). - Execute the fit. The program will output new constants with uncertainties and a correlation matrix. [75]
- In the constants window, set the "Float" option to "yes" for the parameters to be refined (e.g.,
- Iterate: Based on the fit, the simulated spectrum will improve. Assign more lines and refit to further improve accuracy.
Table 2: Key Parameters for High-Resolution IR Spectral Fitting
| Parameter | Symbol | Description | Role in Spectrum Simulation |
|---|---|---|---|
| Band Origin | ν₀ (Origin) | The energy of the pure vibrational transition (Q branch center). | Positions the entire band along the wavenumber axis. |
| Rotational Constant (Ground) | B'' | Relates to the molecular geometry in the ground vibrational state. B = h/(8π²cI) | Governs the spacing of rotational lines in the P and R branches for the ground state. |
| Rotational Constant (Excited) | B' | Relates to the molecular geometry in the excited vibrational state. | Governs the spacing of rotational lines in the P and R branches for the excited state. A difference between B' and B'' creates a gap at the band head. |
| Centrifugal Distortion Constant | D | A small correction for the centrifugal stretching of bonds at high J. | Fine-tunes the positions of high-J lines. |
Machine Learning-Enhanced Simulation
A modern approach to overcoming the computational cost vs. accuracy trade-off in quantum chemistry is the use of machine learning (ML).
Protocol 4: ML-Accelerated IR Simulation with AIQM Models
- Objective: To simulate accurate IR spectra of transition metal complexes with speed接近 semi-empirical methods and accuracy接近 high-level quantum chemistry.
- Principle: Universal ML models like AIQM2 are trained on high-quality reference data (e.g., from coupled-cluster calculations). They learn to predict potential energy surfaces and molecular properties, enabling fast computation of harmonic frequencies with high accuracy. [25]
- Procedure:
- Input Preparation: Generate a reasonable 3D molecular geometry for the transition metal complex.
- Software Setup: Use a computational package that implements the AIQM models, such as
MLatom. - Calculation Execution: Run a geometry optimization followed by a frequency calculation using the AIQM2 Hamiltonian. The calculation typically uses the harmonic approximation.
- Post-Processing: Apply an empirical scaling factor to the computed harmonic frequencies to better match experimental fundamentals. The results include the wavenumber, intensity, and vibrational normal modes for each band. [25]
- Visualization and Assignment: Use the provided tools to visualize the atomic motions associated with each IR-active vibration, aiding in the assignment of spectral features. [25]
The relationship between different computational methods for IR simulation is summarized in the diagram below:
Figure 2: The positioning of ML-enhanced models (AIQM) in the landscape of computational methods for IR simulation, bridging the gap between speed and accuracy.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Materials for Studying Transition Metal Complexes
| Reagent/Material | Function and Application Notes |
|---|---|
| Transition Metal Salts | The source of the metal center (e.g., CoCl₂·6H₂O, Ni(CH₃COO)₂·4H₂O). High-purity salts are used to synthesize complexes. [74] |
| Bidentate Ligands | Molecules with two donor atoms (e.g., amino acids, 2,2'-bipyridine) that form stable chelate complexes, often used to model metal sites in biology. [74] |
| Deuterated Solvents | Used for NMR spectroscopy (e.g., D₂O, DMSO-d₆) to characterize diamagnetic complexes and assess purity. [74] |
| IR Grade KBr | Used to prepare pellets for FT-IR transmission spectroscopy of solid samples. |
| Computational Chemistry Software | Packages for quantum chemical calculations (e.g., for DFT, semi-empirical methods) or specialized spectral simulation (e.g., PGOPHER for gas-phase rotation-vibration spectra). [75] [25] |
Geometry optimization, the process of finding a molecular structure that corresponds to a local minimum on the potential energy surface (PES), represents a fundamental step in computational chemistry workflows for spectroscopic analysis [76]. Within research focused on semi-empirical methods for IR spectra simulation, a robust and well-converged geometry is paramount, as the resulting harmonic frequencies directly depend on the local curvature of the PES at the optimized structure. An incomplete or poorly converged optimization can lead to spurious imaginary frequencies or incorrect vibrational modes, compromising the entire spectroscopic interpretation.
The core challenge lies in the fact that optimizations are iterative procedures that can fail or stall for numerous reasons. This application note addresses the convergence issues specific to geometry optimization, detailing the critical algorithms, thresholds, and practical protocols essential for researchers, particularly those employing semi-empirical methods in drug development.
Core Concepts and Convergence Criteria
A geometry optimization is considered converged when the structure resides in a local minimum on the PES. In practice, this is determined by simultaneously satisfying multiple mathematical criteria related to energy changes, forces (gradients), and atomic displacements [76].
Quantitative Convergence Thresholds
Convergence is typically monitored for up to four quantities: the energy change, the Cartesian gradients, the Cartesian step size, and for periodic systems, the stress energy per atom. The Convergence%Quality keyword offers a convenient way to control these parameters collectively [76]. The following table summarizes the standard threshold values for different quality levels in a typical computational package.
Table 1: Standard Geometry Optimization Convergence Thresholds for Different Quality Settings [76]
| Quality Setting | Energy (Ha) | Gradients (Ha/Å) | Step (Å) | StressEnergyPerAtom (Ha) |
|---|---|---|---|---|
| VeryBasic | 10⁻³ | 10⁻¹ | 1 | 5×10⁻² |
| Basic | 10⁻⁴ | 10⁻² | 0.1 | 5×10⁻³ |
| Normal | 10⁻⁵ | 10⁻³ | 0.01 | 5×10⁻⁴ |
| Good | 10⁻⁶ | 10⁻⁴ | 0.001 | 5×10⁻⁵ |
| VeryGood | 10⁻⁷ | 10⁻⁵ | 0.0001 | 5×10⁻⁶ |
A geometry optimization is considered converged only when all of the following conditions are met [76]:
- The energy change between successive steps is smaller than
Convergence%Energytimes the number of atoms. - The maximum Cartesian nuclear gradient is smaller than
Convergence%Gradient. - The root mean square (RMS) of the Cartesian nuclear gradients is smaller than 2/3 of
Convergence%Gradient. - The maximum Cartesian step is smaller than
Convergence%Step. - The root mean square (RMS) of the Cartesian steps is smaller than 2/3 of
Convergence%Step.
It is crucial to understand that the convergence threshold for coordinates (Convergence%Step) is not a reliable measure for the precision of the final coordinates. For accurate results, one should prioritize tightening the criterion on the gradients rather than on the steps [76].
Optimization Algorithms and Their Characteristics
Optimization algorithms can be broadly divided into two categories: those using Newtonian dynamics with friction and quasi-Newton methods that build an approximation of the Hessian (the matrix of second derivatives) [77].
Table 2: Comparison of Common Local Geometry Optimization Algorithms
| Algorithm | Type | Key Characteristics | Typical Use Case |
|---|---|---|---|
| BFGS | Quasi-Newton | Updates inverse Hessian; often optimal. Good convergence efficiency [77]. | Standard molecular optimizations. |
| L-BFGS | Quasi-Newton | Limited-memory BFGS; low memory footprint [77]. | Large systems (>1000 atoms). |
| FIRE | Dynamics-based | Uses Newtonian dynamics with adaptive friction [77]. | Systems with complex, rough PES. |
| MDMin | Dynamics-based | Modification of velocity-Verlet MD; zeroes momentum when force-momentum dot product is zero [77]. | Early-stage optimization. |
| GPMin | Gaussian Process | Models PES using previous energies/forces; can speed up BFGS [77]. | Expensive energy calculations. |
For semi-empirical methods, which are computationally less expensive, BFGS and its variant L-BFGS are often excellent choices due to their robust and efficient convergence properties.
Troubleshooting Convergence Failures: A Structured Protocol
When a geometry optimization fails to converge, a systematic approach is necessary to diagnose and resolve the issue. The workflow below outlines a standard protocol for troubleshooting.
Figure 1: A workflow for diagnosing and resolving common geometry optimization convergence failures.
Protocol for Resolving Standard Convergence Failures
The following steps provide detailed methodologies corresponding to the troubleshooting workflow in Figure 1.
Step 1: Meticulous Output Analysis Scrutinize the optimization log to identify the unconverged criterion. If the maximum force or step size oscillates without decreasing, the optimizer may be trapped in a flat or rocky region of the PES. Consistent energy increase suggests the optimizer is stepping "uphill," often due to a poor initial Hessian.
Step 2: Initial Geometry Verification Visually inspect the initial molecular structure using a graphical interface. Look for unrealistically short interatomic distances (bad contacts), incorrect stereochemistry, or bond angles that deviate severely from expected values. Even small initial errors can propagate into significant convergence problems. For crystal-based structures, ensure hydrogen atom positions are properly optimized, as they are often inaccurately determined by X-ray diffraction [78].
Step 3: Strategy for Oscillations and Large Steps If the optimization history shows large, oscillating steps without steady progress, temporarily loosen the convergence criteria (e.g., to
Basicquality) [76]. This allows the optimizer to make more significant strides toward the minimum before applying tighter, final convergence thresholds. This can be particularly effective in the initial stages of optimizing a poor starting geometry.Step 4: Strategy for Slow or Stalled Progress When the energy and forces change very slowly, the optimizer may lack a good sense of the PES curvature. The most effective solution is to provide a more accurate initial Hessian. This can be done by using the
CalcFCkeyword, which instructs the program to calculate the Hessian analytically or numerically at the first point, or by reading a Hessian from a previous calculation (ReadFC) [79].Step 5: Strategy for Near-Convergence Failure If the optimization stalls despite being very close to a minimum, slightly tightening the convergence criteria (e.g., to
Goodquality) can often push it to full convergence [76]. Ensure that the computational method (e.g., the quantum chemical engine and its numerical integration grid) is sufficiently accurate to produce noise-free gradients, as numerical noise can prevent tight convergence [76].Step 6: Hierarchical Optimization for Problematic Systems For persistently difficult molecules, a hierarchical optimization strategy is recommended. Begin with a fast, robust method like a semi-empirical (e.g., GFN-xTB [80]) or a low-level DFT calculation with a small basis set and loose thresholds. Once converged, use the resulting geometry as the starting point for a higher-level method, such as a more advanced semi-empirical model or DFT with a larger basis set and tighter thresholds [79]. This multi-step approach efficiently guides the optimization to the correct minimum.
Advanced Techniques: Handling Saddle Points and MECPs
Automatic Restarts from Saddle Points An optimization might converge to a first-order saddle point (transition state) instead of a minimum. This can be identified by a frequency calculation revealing imaginary modes. Some software packages offer an automated restart feature. If a system with disabled symmetry converges to a saddle point, the optimizer can automatically displace the geometry along the imaginary vibrational mode and restart the optimization. This is enabled by setting
MaxRestartsto a value >0 and activating PES point characterization in the properties block [76].Searching for Minimum Energy Crossing Points (MECPs) In photochemistry, exploring intersections between potential energy surfaces of different spin multiplicities is crucial for studying processes like intersystem crossing. The standard algorithm for locating MECPs, as implemented in tools like
sobMECP, involves optimizing along two effective, orthogonal gradients: one that minimizes the energy difference between the two states (f) and another that minimizes the energy of one state while remaining orthogonal to the first (g). The search converges when both the forces and the energy difference are sufficiently small [81].
This section details key software and computational "reagents" essential for conducting research in this field.
Table 3: Key Computational Tools for Geometry Optimization and Analysis
| Tool / Resource | Type | Primary Function in Optimization |
|---|---|---|
| AMS | Software Package | A comprehensive platform with multiple geometry optimizers and sophisticated convergence control [76]. |
| Gaussian | Software Package | A widely used quantum chemistry program supporting various optimization algorithms and keywords like opt=vtight [79]. |
| ASE | Software Library | Provides a Python interface to numerous optimizers (BFGS, FIRE, etc.) and calculators, ideal for scripting custom workflows [77]. |
| GFN-xTB | Semi-empirical Method | A fast, semi-empirical method excellent for hierarchical optimization and pre-optimizing large systems or conformational screening [80]. |
| Multiwfn | Analysis Tool | A versatile wavefunction analyzer for post-optimization tasks like calculating atomic charges (e.g., CHELPG) for background charge setups [78]. |
| sobMECP | Utility | A specialized program for locating minimum energy crossing points between electronic states of different multiplicities [81]. |
| VESTA | Visualization Tool | Used for building and visualizing crystal structures and creating supercells for solid-state calculations [78]. |
In the field of computational spectroscopy, particularly for simulating Infrared (IR) spectra, researchers are constantly faced with a fundamental challenge: reconciling the need for high accuracy with the practical constraints of computational cost and time. This balance is especially critical in industrial applications like drug discovery, where rapid and reliable predictions can significantly accelerate research and development cycles. While high-level ab initio quantum mechanical methods can provide excellent accuracy, their computational expense often makes them prohibitive for large molecules or high-throughput screening. Modern semi-empirical quantum mechanical (SQM) methods, augmented by machine learning (ML) corrections, have emerged as powerful tools that offer a favorable balance, providing sufficient accuracy for many applications at a fraction of the computational cost. This application note details structured protocols and benchmarks to guide researchers in selecting and implementing the most computationally efficient methods for IR spectra simulation without compromising the reliability needed for scientific and drug development purposes.
Accuracy Benchmarking of Computational Methods
Selecting an appropriate method requires a clear understanding of the performance characteristics of different computational approaches. The following tables summarize the accuracy and computational cost of various methods relevant to IR spectra simulation and related properties.
Table 1: Performance Benchmark of Electronic Structure Methods for Key Chemical Properties [82]
| Method Type | Method Name | Conformational Energies | Intermolecular Interactions | Tautomers/Protonation States | Relative Computational Cost |
|---|---|---|---|---|---|
| NDDO-based SQM | PM6 | Moderate | Moderate | Poor | Very Low |
| PM7 | Moderate | Good | Good | Very Low | |
| DFTB-based SQM | GFN2-xTB | Good | Good | Good | Very Low |
| Pure ML Potential | ANI-2x | Good | Good | Poor (for charged species) | Low |
| Hybrid QM/Δ-MLP | AIQM1 | Very Good | Very Good | Very Good | Low |
| QDπ | Very Good | Very Good | Excellent | Low |
Table 2: Performance of Methods for IR Spectroscopy and Reaction Properties [26] [83]
| Method | IR Central Frequency Error | Reaction Energy Error | Transition State Optimization | Barrier Height Error |
|---|---|---|---|---|
| GFN2-xTB | < 10% [83] | Moderate | Feasible | Moderate |
| Common DFT (hybrid/DZ) | ~1-2% | Moderate | Standard | Moderate |
| AIQM2 | N/A | Very Low (Approaches CCSD(T)) | Excellent | Very Low (Approaches CCSD(T)) |
| Gold Standard CCSD(T) | ~0% | Very Low | Computationally Expensive | Very Low |
Detailed Experimental Protocols
Protocol 1: Rapid Generation of Synthetic IR Training Data using Semi-Empirical Methods
This protocol is designed for generating large datasets of synthetic vibrational spectra to train machine learning models, drastically reducing the experimental burden of data collection [84].
Step-by-Step Procedure:
- Curate a Molecular Set: Compile a set of molecular structures of interest. Sources can include public databases like the Worldwide Protein Data Bank (wwPDB). Select molecules based on the availability of 3D geometries.
- Geometry Optimization: For each molecule, perform a geometry optimization using the GFN2-xTB method. Use the
--vtightconvergence criterion (Econv ≤ 5·10⁻⁶ Eh,Gconv ≤ 1·10⁻⁴ Eh·α) to ensure a sufficiently refined structure for frequency calculation [84]. - Frequency Calculation: Using the optimized geometry, calculate the vibrational frequencies and intensities with the GFN2-xTB method, typically using the
--ohessoption in thextbpackage [84]. - Quality Control: Parse the output files to check for the presence of imaginary frequencies. Reject any molecules that exhibit imaginary modes, as these indicate the structure is not at a true energy minimum [84].
- Spectral Broadening: Convert the calculated line spectra (stick diagrams) into continuous, realistic spectra by applying a Voigt profile broadening. This profile combines Lorentzian (intrinsic line shape) and Gaussian (instrumental, Doppler effects) broadening [84].
- Artifact Introduction (Optional): To improve the model's robustness, artificially introduce realistic artifacts into the spectra, such as:
- Additive Gaussian noise.
- Fluorescent background signals.
- Baseline variations [84].
The following workflow diagram illustrates this protocol:
Protocol 2: Machine Learning-Assisted Real-Time Yield Prediction from Inline IR
This protocol leverages a machine learning model, trained on synthetic spectra, to predict reaction yields in real-time during automated synthesis, enabling closed-loop optimization [85].
Step-by-Step Procedure:
- Acquire Reference Spectra: Measure the FTIR spectra of pure starting materials and the expected pure product under reaction conditions. Repeat measurements to account for instrumental noise.
- Generate Training Data via Linear Combination: Create a large training dataset by simulating the spectra of reaction mixtures. This is done by taking linear combinations of the pure component spectra, weighted by a virtual percent yield (e.g., from 0% to 100%) and a simulated decomposition rate.
- Formula:
Spectrum_mixture = (1 - cyield) * Spectrum_reactants + cyield * Spectrum_product[85].
- Formula:
- Preprocess Spectral Data: Differentiate the spectra (e.g., using first or second derivatives) to enhance subtle spectral features. Restrict the analysis to the most informative regions, such as the fingerprint region (699–1692 cm⁻¹), to improve model performance [85].
- Train a Neural Network Model: Construct and train a one-dimensional convolutional neural network (1D-CNN) or a similar deep learning model. Use the simulated spectra as input (
X) and the virtual percent yield (cyield) as the target output. - Integrate Model for Real-Time Prediction: Deploy the trained model within an automated flow chemistry system. The system should consist of:
- A flow reactor.
- An inline FTIR spectrometer for continuous measurement.
- A programmable logic controller (PLC) to control pumps and heaters based on model predictions [85].
- Closed-Loop Optimization: Use the real-time yield predictions as feedback for a Bayesian optimization algorithm. The algorithm automatically adjusts reaction parameters (e.g., flow rate, temperature) to maximize the predicted yield [85].
Protocol 3: Transfer Learning for Raman Spectral Classification with Limited Data
This protocol uses synthetic data from Protocol 1 to pre-train a deep learning model, which is then fine-tuned on a small set of experimental Raman spectra, overcoming data scarcity [84].
Step-by-Step Procedure:
- Generate Large Synthetic Pretraining Set: Follow Protocol 1 to generate thousands of synthetic vibrational (Raman) spectra for a diverse set of molecules.
- Pretrain a Deep Learning Model: Use the synthetic spectra to pretrain a 1D-CNN model. The model learns to extract general, low-level spectral features (e.g., peak shapes, bandwidths) from this data-rich source domain.
- Acquire Limited Target Experimental Data: Collect a smaller, task-specific dataset of experimental Raman spectra (e.g., spectra from different bacterial species).
- Fine-Tune the Pretrained Model: Replace the final classification layer of the pretrained CNN. Then, train (fine-tune) the network on the small experimental dataset. This process adapts the general model to the specific patterns and noise characteristics of the target domain.
- Validate Model Performance: Evaluate the fine-tuned model on a held-out test set of experimental spectra to assess its classification accuracy and generalizability.
The logical relationship of this transfer learning approach is shown below:
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Key Software Tools for Cost-Effective IR Spectra Simulation
| Tool Name | Type | Primary Function | Key Advantage |
|---|---|---|---|
| GFN2-xTB [83] [84] | Semi-empirical QM | Geometry optimization, frequency calculation, NCI | Excellent speed/accuracy trade-off; suitable for large systems. |
| MLatom [26] | AI-Enhanced QM Platform | Runs AIQM1, AIQM2, and other ML-potentials | Provides coupled-cluster level accuracy at SQM cost. |
| xtb [84] | Software Package | Provides GFN2-xTB and related methods | Open-source, user-friendly, and efficient. |
| MOPAC [82] | Software Package | Provides PM6, PM7, and other SQM methods | Well-established platform for SQM calculations. |
| DeePMD-kit [82] | Machine Learning Potentials | Runs DPRc and other deep neural network potentials | Enables ML-based molecular dynamics simulations. |
| ANI-2x [82] | Pure ML Potential | Molecular energy and force prediction | High accuracy for neutral organic molecules; very fast. |
Benchmarking Performance: Validation Strategies and Method Comparisons
Within the broader context of research on semi-empirical methods for infrared (IR) spectra simulation, experimental validation using known pharmaceutical compounds remains a critical step for assessing the accuracy and reliability of computational approaches. As IR spectroscopy continues to be a vital tool for identifying functional groups and characterizing molecular structures in drug development, establishing robust protocols for validating simulated spectra against experimental data is paramount for researchers and pharmaceutical scientists [3] [86]. This application note details specific case studies and methodologies for the experimental validation of IR spectra simulation techniques, providing structured protocols and quantitative comparisons to guide research in this field.
Computational Methods for IR Spectra Simulation
The simulation of IR spectra can be approached through various computational methods, each with distinct advantages and limitations. Understanding this methodological landscape is essential for selecting appropriate validation strategies.
- Semi-Empirical Methods: These approaches, such as the Austin Model 1 (AM1) and Zerner's Intermediate Neglect of Differential Overlap (ZINDO), parameterize certain quantum chemical integrals based on experimental data. They offer a balance between computational cost and accuracy, making them suitable for initial screening and studies of large molecular systems like graphene quantum dots or pharmaceutical fragments [3]. The AM1 method, for instance, has been shown to successfully predict infrared intensities and spectral patterns for complex organic materials.
- Density Functional Theory (DFT): DFT methods, particularly those using hybrid functionals like B3LYP, generally provide higher accuracy than semi-empirical methods for vibrational frequency calculations. Their performance is highly dependent on the selection of the basis set, with the inclusion of polarization functions being critical for reliable results [87] [10] [28].
- Novel Multi-Molecular Approaches: Traditional simulations often use single-molecule models in a vacuum, which can lead to inaccuracies, especially for molecules with intermolecular interactions like hydrogen bonding. Recent advances propose multi-molecular fragment models that better mimic the molecular environment found in solid-state pharmaceuticals, significantly improving agreement with experimental data [86].
Case Studies in Pharmaceutical Validation
Bosentan Hydrate and Sildenafil Base Binary System
A compelling validation case study involves the investigation of interactions between bosentan monohydrate (BOS) and sildenafil base (SIL) induced by high-energy ball milling [87].
Experimental Protocol:
- Sample Preparation: Prepare physical mixtures and co-milled binary solid dispersions of BOS and SIL using a high-energy ball mill (e.g., Pulverisette 7, Fritsch) with zirconium oxide jars and milling balls.
- FT-IR Spectroscopy: Acquire infrared spectra of single components, physical mixtures, and co-milled formulations using a standard FT-IR spectrometer.
- DFT Calculations: Perform theoretical calculations for sildenafil isomers and for bosentan trimers/tetramers to interpret the experimental spectra. Geometry optimization and frequency calculations are typically conducted using a suitable DFT functional.
- Spectral Analysis: Compare experimental and theoretical spectra, focusing on shifts in wavenumbers related to hydrogen bonding and molecular rearrangements. Use classification methods to distinguish between amorphous solid dispersions and physical mixtures based on their unique vibrational signatures.
Key Findings: The study successfully used DFT calculations to interpret the IR spectra, revealing that the amorphous form of the BOS-SIL dispersion was stabilized by hydrogen bonds between the two drug molecules. Significant changes in vibrational dynamics due to the amorphization of crystalline drugs were observed and theoretically validated [87].
Finasteride, Lamivudine, and Repaglinide via Multi-Molecular Fragment Method
This case study validates a novel multi-molecular fragmentation method for simulating the harmonic vibrational frequencies of three pharmaceutical molecules: Finasteride (FIN), Lamivudine (LAM), and Repaglinide (REP) [86].
Experimental Protocol:
- Model Construction:
- Single-Molecular Model: The isolated drug molecule optimized in a vacuum.
- Central-Molecular Model: A single molecule optimized within a fixed crystal environment to account for spatial hindrance.
- Multi-Molecular Fragment Model: A cluster of several molecules extracted from the crystal structure to capture intermolecular interactions like hydrogen bonding.
- Computational Details: Simulations are performed using Generalized Gradient Approximations (GGA) with the PBE functional within materials studio software.
- Experimental Comparison: Theoretical vibrational frequencies are compared against experimental FT-IR data.
- Model Construction:
Key Findings: The multi-molecular fragment method demonstrated superior agreement with experimental results compared to traditional single-molecule calculations and even the central-molecular model. The following table summarizes the quantitative validation data [86]:
Table 1: Performance of Computational Models for Pharmaceutical Molecules
| Pharmaceutical Molecule | Computational Model | Mean Absolute Error (MAE, cm⁻¹) | Root Mean Squared Error (RMSE, cm⁻¹) |
|---|---|---|---|
| Finasteride (FIN) | Single-Molecular (Scaled) | Not Specified | Highest among models |
| Central-Molecular | Reduced vs. Single-Model | Reduced vs. Single-Model | |
| Multi-Molecular Fragment | 8.21 | 18.35 | |
| Lamivudine (LAM) | Single-Molecular (Scaled) | Not Specified | Highest among models |
| Central-Molecular | Reduced vs. Single-Model | Reduced vs. Single-Model | |
| Multi-Molecular Fragment | 15.95 | 26.46 | |
| Repaglinide (REP) | Single-Molecular (Scaled) | Not Specified | Highest among models |
| Central-Molecular | Reduced vs. Single-Model | Reduced vs. Single-Model | |
| Multi-Molecular Fragment | 12.10 | 25.82 |
The data conclusively shows that the multi-molecular fragment interception method provides the most accurate simulation, with the lowest MAE and RMSE values across all three model pharmaceuticals.
Experimental Protocol for Validation
This section provides a detailed, step-by-step protocol for the experimental validation of simulated IR spectra based on the cited case studies.
Workflow for Validation
The following diagram illustrates the logical workflow for the validation of simulated IR spectra against experimental data.
Step-by-Step Procedure
- Select Pharmaceutical Compound: Choose a known, well-characterized drug substance for which a high-purity sample is available. Examples include Finasteride, Lamivudine, or Repaglinide [86].
- Obtain Experimental IR Spectrum:
- Prepare a sample of the compound using a suitable technique (e.g., KBr pellet for solids).
- Acquire the FT-IR spectrum using a calibrated spectrometer. Record the spectrum across a relevant wavenumber range (e.g., 400-4000 cm⁻¹) with a resolution of at least 4 cm⁻¹.
- Document all sample preparation and instrument parameters meticulously.
- Choose Computational Method(s):
- Construct Molecular Model(s):
- Single-Molecule Model: Build an isolated molecule based on its covalent structure.
- Multi-Molecular Model (Recommended): If crystal structure data is available, construct a cluster of molecules to capture intermolecular interactions. This is crucial for accurately simulating hydrogen-bonded functional groups [86].
- Perform Geometry Optimization: Run a geometry optimization calculation to find the most stable energy conformation of the constructed model(s) before frequency calculation. The convergence limit for the energy gradient should be set stringently (e.g., 0.05 kcal/mol/Å) [3].
- Calculate Vibrational Frequencies: Using the optimized geometry, perform a frequency calculation to obtain the theoretical harmonic vibrational frequencies, IR intensities, and, if applicable, Raman activities.
- Compare and Analyze Spectra:
- Visually overlay the simulated spectrum with the experimental one.
- Identify key functional group regions (e.g., C=O stretch ~1700 cm⁻¹, O-H stretch ~3000-3500 cm⁻¹) and note the presence or absence of corresponding peaks, as well as any significant shifts [3] [87].
- Pay particular attention to the fingerprint region (400-1500 cm⁻¹), which is highly sensitive to molecular structure and environment.
- Quantitative Error Assessment:
- Calculate quantitative error metrics such as Mean Absolute Error (MAE) and Root Mean Squared Error (RMSE) for the vibrational frequencies of key peaks compared to experimental values [86].
- A well-validated method should have low MAE and RMSE values, indicating close agreement with experiment.
The Scientist's Toolkit
The following table details essential research reagents, software, and computational solutions used in the featured experiments for IR spectra simulation and validation.
Table 2: Essential Research Reagents and Solutions for IR Spectral Validation
| Item Name | Type/Example | Function in Validation |
|---|---|---|
| High-Purity Pharmaceutical Standards | Finasteride, Lamivudine, Repaglinide, Bosentan Monohydrate, Sildenafil Base | Serve as the benchmark compounds for acquiring experimental FT-IR spectra to validate computational simulations. |
| FT-IR Spectrometer | Fourier Transform Infrared Spectrometer | Measures the experimental infrared absorption spectrum of the sample, providing the fundamental data for validation. |
| Computational Chemistry Software | Gaussian09, Material Studio, Winmostar | Provides the platform to run semi-empirical, DFT, and other quantum mechanical calculations for geometry optimization and vibrational frequency simulation [88] [86]. |
| Semi-Empirical Methods | AM1, ZINDO | Offer a computationally efficient method for simulating IR spectra and electronic transitions, useful for large systems or initial screening [3]. |
| Density Functional Theory (DFT) | PBE, B3LYP-D3(BJ) | Provides higher-accuracy simulations of vibrational frequencies and intensities when combined with an appropriate basis set [86] [28]. |
| Spectral Database | NIST IR Database, LIDA, KnowItAll | Provides reference experimental spectra for comparison and for training machine learning models [89] [90] [28]. |
The experimental validation of simulated IR spectra using known pharmaceutical compounds is a critical process for establishing confidence in computational methods. Case studies involving drugs like bosentan-sildenafil mixtures and finasteride demonstrate that while traditional single-molecule simulations have limitations, advanced approaches like multi-molecular fragmentation and careful DFT calculations can achieve remarkable agreement with experimental data. The provided protocols, workflows, and toolkit offer researchers a structured framework for conducting rigorous validations, thereby strengthening the role of semi-empirical and other computational methods in the modern drug development pipeline.
Infrared (IR) spectroscopy is a cornerstone analytical technique for molecular structure identification, playing a critical role in chemistry, materials science, and drug discovery [91] [92]. For theoretical simulation of IR spectra, Density Functional Theory (DFT) has long been the gold standard for its high accuracy, but its prohibitive computational cost limits practical application in high-throughput settings [92]. Semi-empirical methods offer faster calculations but often suffer from insufficient accuracy [33] [58].
The emergence of Machine Learning (ML) methods provides a transformative solution, bridging the gap between these two approaches. This application note quantitatively compares the accuracy and computational efficiency of modern ML-based approaches against traditional DFT and semi-empirical methods for IR spectra simulation, providing clear protocols for researchers.
Quantitative Comparison of Methods
The table below summarizes the key performance metrics of different computational methods for IR spectra simulation, based on recent literature.
Table 1: Performance Comparison of Methods for IR Spectra Simulation
| Method | Typical Accuracy (vs. Experiment) | Computational Cost | Key Advantages | Primary Limitations |
|---|---|---|---|---|
| DFT (AIMD) | High (Gold standard) | Extremely High | Naturally includes anharmonicity and temperature effects [91]. | Computationally prohibitive for large systems/long timescales [91] [57]. |
| MLIPs (e.g., MACE4IR, PALIRS) | High (Close to DFT) [57] | Low (Orders of magnitude faster than AIMD) [91] | Near-DFT accuracy at semi-empirical speed; captures anharmonicity [91] [57]. | Requires quality training data; foundation models reduce but do not eliminate this need [91] [57]. |
| Semi-Empirical (e.g., GFN2-xTB) | Moderate [33] [58] | Low | Very fast calculation speed [33]. | Quantitative discrepancies and limited accuracy [33] [58]. |
| Harmonic Approximation (DFT) | Moderate (Neglects anharmonicity) [91] | Medium | Computationally efficient for frequency analysis [57]. | Neglects anharmonic and temperature effects, leading inaccuracies [91] [93]. |
Performance of Machine Learning Interatomic Potentials (MLIPs)
MLIPs demonstrate remarkable performance in predicting key quantum mechanical properties necessary for IR spectra, as shown by the foundation model MACE4IR trained on a massive DFT dataset.
Table 2: Accuracy of the MACE4IR Foundation Model on Key Properties [57]
| Training Set Size | Energy MAE (meV/atom) | Force MAE (meV/Å) | Dipole Moment MAE (meÅ) |
|---|---|---|---|
| 1 Million Geometries | 5.1 | 62.7 | 26.3 |
| 7 Million+ Geometries | 3.8 | 50.1 | 25.3 |
These accurate predictions of forces and dipole moments enable the calculation of IR spectra that agree well with both theoretical DFT references and experimental data at a fraction of the computational cost of AIMD [57]. The PALIRS framework demonstrates that MLIPs can reproduce AIMD-quality IR spectra for organic molecules while reducing computational cost by several orders of magnitude [91].
Detailed Protocols for ML-Enhanced IR Simulation
This section outlines two distinct protocols for implementing ML-enhanced IR spectra simulation.
Protocol 1: Active Learning Workflow with PALIRS
The PALIRS framework employs an active learning cycle to efficiently build accurate, system-specific MLIPs [91].
Objective: To construct a high-quality dataset and train an MLIP for accurate IR spectra prediction of specific molecular systems with minimal computational expense. Key Components:
- Software: PALIRS open-source package [91].
- MLIP Architecture: An ensemble of three MACE models for energy, force, and uncertainty prediction [91].
- Dipole Moment Model: A separate MACE model trained specifically for dipole moment prediction [91].
Step-by-Step Procedure:
- Initial Dataset Generation: Generate initial molecular geometries by sampling along normal vibrational modes derived from DFT calculations (e.g., using FHI-aims) [91].
- Initial MLIP Training: Train the initial MLIP ensemble on this small dataset (~2000 structures) [91].
- Active Learning Cycle: a. MLMD Simulation: Run molecular dynamics simulations using the current MLIP at multiple temperatures (e.g., 300 K, 500 K, 700 K) to explore configuration space [91]. b. Uncertainty Quantification: Use the MLIP ensemble to identify configurations with the highest uncertainty in force predictions [91]. c. DFT Calculation: Perform targeted DFT calculations on these high-uncertainty configurations to obtain accurate energies, forces, and dipole moments [91]. d. Dataset Expansion & Retraining: Add the new data to the training set and retrain the MLIP ensemble [91]. e. Iteration: Repeat steps a-d until convergence (e.g., for ~40 cycles, resulting in a final dataset of ~16,000 structures) [91].
- Dipole Moment Model Training: After the MLIP is converged, train a separate ML model on the final dataset to predict dipole moments [91].
- Production MLMD and Spectrum Calculation: Run a long, production-scale MLMD simulation. Use the dipole moment model to compute the dipole time series and calculate the IR spectrum via the Fourier transform of its autocorrelation function [91].
The following diagram illustrates this iterative workflow:
Protocol 2: Utilizing a Foundation Model (MACE4IR)
For a broader range of molecules, using a pre-trained foundation model like MACE4IR offers a direct path to spectra without system-specific training [57].
Objective: To rapidly predict IR spectra for diverse molecules using a general-purpose, pre-trained ML model. Key Components:
- Software: The pre-trained MACE4IR model, which includes two MACE models for energies/forces and dipole moments [57].
- Input: Molecular geometry.
Step-by-Step Procedure:
- Model Acquisition: Obtain the pre-trained MACE4IR model.
- Structure Preparation: Provide the 3D molecular structure of the compound of interest.
- Spectrum Calculation:
- Option A (Harmonic): Directly compute the harmonic vibrational frequencies and intensities for a rapid estimate [57].
- Option B (MD-based): Run an MLMD simulation using the model's MLIP, compute dipole moments along the trajectory, and generate the spectrum via the dipole autocorrelation function to include anharmonic and temperature effects [57].
- Analysis: Compare the predicted spectrum to experimental data or use it for spectral assignment.
The Scientist's Toolkit: Essential Research Reagents
The table below lists key software and methodological "reagents" essential for implementing the aforementioned protocols.
Table 3: Key Research Reagents for ML-Enhanced IR Spectroscopy
| Tool / Resource | Type | Function in Protocol | Key Features |
|---|---|---|---|
| PALIRS [91] | Software Package | Protocol 1: Active Learning Workflow | Open-source Python code for active learning-enhanced MLIP training and IR spectra prediction. |
| MACE4IR [57] | Foundation Model | Protocol 2: Direct Prediction | Pre-trained ML model for accurate energy, force, and dipole moment prediction across ~80 elements. |
| MACE Architecture [91] [57] | ML Model Architecture | Core component of both protocols | A message-passing neural network that forms the backbone of both PALIRS and MACE4IR. |
| AIQM Models [33] [58] | ML Model Series | Alternative ML Approach | ML models targeting CCSD(T)-level accuracy for IR spectra with speed near semi-empirical methods. |
| MLatom [33] [58] | Software Package | Platform for AIQM and other ML calculations | Provides implementations for calculating IR spectra with AIQM, DFT, and other quantum mechanical methods. |
| Active Learning Sampling | Methodological Strategy | Protocol 1: Data Curation | Strategy to selectively run DFT on high-uncertainty configurations, maximizing data efficiency [91]. |
Semi-empirical quantum chemistry methods provide a crucial balance between computational cost and accuracy, making them indispensable for simulating large molecular systems and high-throughput calculations in research areas such as drug development and materials science [6] [94]. Among these methods, AM1, PM6, and PM7 represent significant developments in the parameterization of Hamiltonian models, each offering distinct advantages and limitations. This application note provides a structured benchmark of these three methods, delivering quantitative performance data and detailed protocols to guide researchers in selecting and applying the most appropriate method for their specific molecular systems, with a particular focus on IR spectra simulation.
Performance Benchmarks and Quantitative Comparison
Accuracy in Energetic Properties
The performance of AM1, PM6, and PM7 varies significantly across different chemical systems. Quantitative benchmarks against higher-level theoretical calculations provide clear guidance for method selection.
Table 1: Mean Unsigned Error (MUE, kJ/mol) for Isolated Proton Transfer Reactions [95]
| Chemical Group | AM1 | PM6 | PM7 |
|---|---|---|---|
| –NH3 (Ammonium) | 42.9 | 15.7 | 13.0 |
| COOH (Carboxyl) | 38.7 | 22.7 | 10.3 |
| +CNH2 (Guanidinium) | 26.2 | 25.4 | 14.1 |
| NH (Amide) | 28.3 | 16.1 | 7.03 |
| PhOH (Phenol) | 44.3 | 15.2 | 10.2 |
| Q (Quinone) | 52.3 | 23.1 | 14.1 |
| –SH (Thiol) | 29.5 | 24.2 | 27.6 |
| H2O (Water) | 23.2 | 18.2 | 15.7 |
| Average | 35.0 | 20.3 | 13.4 |
For soot formation simulations involving polycyclic aromatic hydrocarbons (PAHs) with 4 to 24 carbon atoms, the energy profiles along molecular dynamics trajectories were qualitatively correct for all three methods, though none provided quantitatively accurate thermodynamic or kinetic data [6]. In these systems, AM1 surprisingly outperformed PM6 and PM7 in energy profile similarity, though GFN2-xTB and DFTB3 methods showed superior overall performance [6].
Geometries and Non-Covalent Interactions
For ground-state properties including geometries, the OMx methods generally show the best performance among semiempirical methods, with PM6 and PM7 being the best among MNDO-type methods [9]. PM7 includes specific corrections for intermolecular dispersion and hydrogen bond interactions, making it particularly suitable for systems where non-covalent interactions play a critical role [6] [94].
In benchmark studies evaluating noncovalent interactions, PM7 demonstrates comparable accuracy to OMx-Dn methods for small complexes, with particular strength in modeling hydrogen-bonded complexes due to its explicit hydrogen-bond corrections [9]. However, for very large complexes, the OMx-Dn methods are superior [9].
Detailed Experimental Protocols
General Workflow for IR Spectra Simulation
Figure 1: Computational workflow for simulating IR spectra using semi-empirical methods
Protocol 1: IR Spectra Simulation for Functionalized Graphene Quantum Dots
This protocol is adapted from studies on pyrene-like molecules as models for functionalized graphene quantum dots (GQDs) [3].
Initial Structure Construction
- Build the initial molecular structure of the pyrene-like GQD using standard bond lengths (CC: 1.397 Å, CH: 1.084 Å) and bond angles (120°) for polycyclic aromatic hydrocarbons [3].
Geometry Optimization
- Perform initial optimization using molecular mechanics to generate a reasonable starting geometry [3].
- Conduct semi-empirical re-optimization using the AM1 method with the following parameters [3]:
- Convergence limit: gradient of energy change ≤ 0.05 kcal/mol/Ångstrom
- Optimization algorithm: second-order Taylor energy expansion around the current point
- This optimization is performed with the Winmostar package (or equivalent software)
Vibrational Frequency Calculation
- Execute a force calculation on the optimized structure to obtain vibrational modes [3].
- Calculate IR intensities for each vibrational mode.
Spectral Analysis
Protocol 2: High-Throughput Reaction Sampling for Soot Formation
This protocol is designed for massive reaction event sampling in soot formation processes [6].
System Preparation
- Construct hydrocarbon compounds and radicals relevant to soot precursors (4-24 carbon atoms)
- Include diverse reaction types: H-abstraction, acetylene addition, radical recombination
Molecular Dynamics Simulations
Energy Profile Validation
- Extract potential energy along MD trajectories
- Compare with benchmark DFT calculations (M06-2x/def2TZVPP) using statistical indicators [6]:
- Maximum unsigned deviation (MAX)
- Regularized relative RMSE
Application Guidelines
Protocol 3: Proton Transfer Reactions in Biochemical Systems
This protocol is optimized for simulating proton transfer reactions relevant to enzymatic mechanisms and drug design [95].
System Setup
- Select proton transfer reactions representing biologically relevant chemical groups:
- Amino acid side chains (-NH₃, COOH, -SH)
- Functional groups in nucleobases
- Water molecules and hydroxyl groups
- Select proton transfer reactions representing biologically relevant chemical groups:
Single-Point Energy Calculations
- Perform geometry optimization of reactant, product, and transition state structures
- Calculate relative energies for proton transfer reactions
- Compare with MP2/def2-TZVP reference data [95]
Performance Validation
- Compute mean unsigned errors for each chemical group
- For systems with large errors (>20 kJ/mol), consider method-specific parametrization or higher-level methods
The Scientist's Toolkit
Table 2: Essential Computational Tools for Semi-Empirical Methods
| Tool/Resource | Function | Application Context |
|---|---|---|
| Winmostar | Software package for geometry optimization and vibrational frequency calculation with AM1 method | IR spectra simulation of graphene quantum dots and organic molecules [3] |
| MLatom | Platform implementing AIQM models and various semi-empirical methods for IR spectra calculation | Machine-learning enhanced spectral simulation with interpretation of vibrational modes [33] [58] |
| GFN2-xTB | Semi-empirical tight-binding method with D4 dispersion | High-accuracy alternative for systems where PM7 underperforms [6] [95] |
| QM/MM Packages | Hybrid quantum mechanics/molecular mechanics simulation environments | Modeling proton transfer reactions in microsolvated and enzymatic environments [95] |
The benchmark data presented in this application note demonstrates that PM7 generally provides the best performance among the three methods for most molecular systems, particularly for proton transfer reactions and non-covalent interactions. However, AM1 shows specific utility for IR spectra simulation of graphene-based systems, while PM6 represents a balanced option for general organic molecules. Researchers should select methods based on their specific chemical systems and accuracy requirements, using the provided protocols to ensure reliable implementation. For future research, machine-learning corrected approaches such as PM6-ML show promise for significantly improving accuracy while maintaining computational efficiency [95].
The accurate simulation of molecular properties and reaction mechanisms is a cornerstone of modern computational chemistry, with particular importance in fields such as drug design and materials science. For decades, computational chemists have faced a persistent challenge: the trade-off between the high accuracy of quantum mechanical (QM) methods and their prohibitive computational cost. While the coupled cluster with single, double, and perturbative triple excitations (CCSD(T)) method is regarded as the "gold standard" for accuracy, its application remains limited to relatively small systems due to computational scaling that increases with the seventh power of the number of atoms [26] [96].
Semi-empirical quantum mechanical methods offer significantly faster computation but with substantially reduced accuracy, creating a gap between what is computationally feasible and what is scientifically desirable. The emergence of artificial intelligence (AI) and machine learning (ML) has introduced a paradigm shift in this landscape, enabling the development of hybrid approaches that combine the speed of semi-empirical methods with the accuracy of high-level quantum chemistry [97] [98].
This application note focuses on two groundbreaking ML-enhanced quantum mechanical methods: AIQM1 and AIQM2. These methods represent significant advancements in computational chemistry, approaching CCSD(T)-level accuracy while maintaining computational costs comparable to fast semi-empirical methods. Specifically within the context of IR spectra simulation research, these methods enable the accurate prediction of molecular vibrational properties that were previously computationally prohibitive at this accuracy level [24] [58].
Methodological Foundations
AIQM1 Architecture and Implementation
The AIQM1 (artificial intelligence–quantum mechanical method 1) framework establishes the foundational architecture for AI-enhanced quantum chemical methods. Its design incorporates three distinct components that work synergistically to achieve high accuracy at low computational cost [97].
The total energy in AIQM1 is calculated as the sum of three contributions:
[E{\text{AIQM1}} = E{\text{SQM}} + E{\text{NN}} + E{\text{disp}}]
Where (E{\text{SQM}}) represents the energy from a semiempirical quantum mechanical method, (E{\text{NN}}) is the neural network correction, and (E_{\text{disp}}) represents dispersion corrections [97].
For its baseline SQM component, AIQM1 utilizes a modified version of the orthogonalization- and dispersion-corrected method 2 (ODM2) Hamiltonian, referred to as ODM2*, with the original dispersion corrections removed. The neural network component employs an ANI-type potential, summing atomic contributions from individual atoms within a defined cutoff radius. This architecture uses the Δ-learning approach, where the neural network learns the difference between low-level SQM and high-level target methods rather than learning the potential energy surface directly. Finally, state-of-the-art D4 dispersion corrections including Axilrod–Teller–Muto three-body contributions are added to properly describe long-range noncovalent interactions [97].
The training of AIQM1 utilized the ANI-1x and ANI-1ccx datasets, which contain millions of geometries of small neutral, closed-shell molecules with up to 8 non-hydrogen atoms (H, C, N, O elements). The neural network weights were obtained through a two-step process: first, fitting to differences between ωB97X/def2-TZVP and ODM2* potentials, followed by refinement using transfer learning to approach CCSD(T)*/CBS accuracy [97].
AIQM2 Architectural Advancements
Building upon the success of AIQM1, the AIQM2 method introduces several key architectural improvements that enhance both accuracy and applicability, particularly for reaction simulations [26] [99].
AIQM2 modifies the baseline method, replacing ODM2* with GFN2-xTB* (GFN2-xTB with D4 dispersion corrections removed), leveraging its broader applicability in reaction exploration. The energy expression for AIQM2 is:
[E{\text{AIQM2}} = E{\text{GFN2-xTB*}} + E{\text{NN}} + E{\text{D4}(\omega\text{B97X})}]
The neural network component consists of an ensemble of 8 ANI neural networks that correct the baseline GFN2-xTB* energy. The dispersion correction uses the D4 model parameterized for the ωB97X functional. This revised architecture enables AIQM2 to achieve superior performance for transition state optimizations and barrier heights, addressing limitations observed in AIQM1 and other universal interatomic potentials [26].
A critical advantage of AIQM2 is its provision of uncertainty estimates through the neural network ensemble, allowing users to assess the reliability of predictions—a feature particularly valuable when exploring new chemical spaces [26].
Table 1: Comparison of AIQM1 and AIQM2 Method Architectures
| Component | AIQM1 | AIQM2 |
|---|---|---|
| Baseline SQM Method | ODM2* (modified ODM2) | GFN2-xTB* (GFN2-xTB with D4 removed) |
| Neural Network Correction | ANI-type potential (atomic contributions) | Ensemble of 8 ANI neural networks |
| Dispersion Correction | D4 with Axilrod–Teller–Muto three-body terms | D4(ωB97X) |
| Δ-Learning Target | CCSD(T)*/CBS | DLPNO-CCSD(T)/CBS |
| Uncertainty Estimation | Not available | Available via neural network ensemble |
Workflow Diagram
Diagram 1: AIQM1/AIQM2 Computational Workflow. The diagram illustrates the sequential integration of semiempirical quantum mechanical (SQM) calculation, neural network correction, and dispersion correction to generate final electronic properties.
Performance Benchmarks and Accuracy Assessment
Accuracy Across Chemical Properties
Extensive benchmarking against established datasets reveals that both AIQM1 and AIQM2 achieve remarkable accuracy across diverse molecular properties, often approaching or exceeding CCSD(T) quality while maintaining computational efficiency far beyond traditional quantum chemical methods [97] [26].
AIQM1 demonstrates exceptional performance for ground-state energies of diverse organic compounds, with accuracy closely matching coupled cluster methods. The method successfully predicts geometries for challenging systems such as large conjugated compounds, with results for fullerene C60 aligning closely with experimental data. Notably, AIQM1 maintains good accuracy for ionic and excited-state properties despite never being explicitly fitted to these properties during training [97].
AIQM2 shows significant improvements particularly in reaction energies, transition state optimizations, and barrier heights—areas where AIQM1 exhibited limitations. For reaction energies of large systems and certain non-covalent interactions, AIQM2 outperforms hybrid and double-hybrid DFT methods with common double-ζ basis sets. In reaction dynamics simulations, AIQM2 enables the propagation of thousands of high-quality trajectories overnight, achieving product distributions at nearly coupled-cluster level accuracy [26].
Table 2: Performance Benchmarks for AIQM Methods (Mean Absolute Errors in kcal/mol)
| Property Type | AIQM1 | AIQM2 | Typical DFT | Target CCSD(T) |
|---|---|---|---|---|
| Ground-State Energies | ~1.0 [97] | ~1.0 [26] | 1.0-5.0 [96] | 0.0 (reference) |
| Reaction Energies | Subpar [26] | <1.0 [26] | 1.26-5.26 [96] | 0.0 (reference) |
| Barrier Heights | Subpar [26] | <1.0 [26] | 1.50-4.22 [96] | 0.0 (reference) |
| Non-Covalent Interactions | Good [97] | Excellent [26] | Varies widely [100] | 0.0 (reference) |
| Geometries | Close to experiment [97] | Improved [26] | Good [101] | Excellent |
Computational Efficiency
The computational efficiency of AIQM methods represents one of their most transformative characteristics. AIQM1 achieves computational speeds comparable to approximate low-level semiempirical QM methods while approaching the accuracy of the coupled cluster gold standard [97].
AIQM2 demonstrates even more impressive performance, with speeds orders of magnitude faster than common DFT methods while maintaining superior accuracy. This efficiency enables previously infeasible computational experiments, such as extensive reaction dynamics studies that can be completed overnight on limited computational resources [26] [99].
In comparative studies, hybrid approaches using GFN-xTB methods for geometry optimization with higher-level single-point energy corrections have shown 50-fold reductions in computational time while achieving DFT-D3-level accuracy [101]. This aligns with the performance profile of AIQM methods, which leverage similar principles of multi-level integration but with more sophisticated ML-driven corrections.
Application to IR Spectroscopy
Theoretical Background
Infrared (IR) spectroscopy serves as a powerful analytical tool for identifying molecular structures and studying chemical properties. Theoretical approaches to IR spectrum simulation have traditionally relied on quantum chemical calculations of molecular vibrational frequencies and intensities, with accuracy directly correlated with computational cost [24].
The harmonic approximation provides the foundation for most computational IR spectroscopy, where vibrational frequencies are calculated from the second derivatives of the potential energy surface at the equilibrium geometry. While this approximation works well for many systems, anharmonic effects can be significant for certain molecular types and vibrational modes [24].
Density functional theory has served as the workhorse for computational IR spectroscopy, but its accuracy is limited by functional choice and basis set size. Higher-level methods like CCSD(T) provide superior accuracy but remain computationally prohibitive for all but the smallest systems [24] [58].
AIQM Implementation for IR Spectra
The application of AIQM models to IR spectroscopy represents a breakthrough in achieving accurate spectral predictions with computational efficiency previously unattainable with traditional QM methods [24] [58].
The implementation utilizes AIQM methods to calculate the harmonic force constants through analytical second derivatives of the energy with respect to nuclear coordinates. For AIQM2, the Hessian matrix is computed as the sum of three contributions: numerical differentiation of analytical gradients from GFN2-xTB*, analytical Hessians from the neural network correction, and numerical differentiation of D4 dispersion corrections [99].
To account for systematic errors in the harmonic approximation, frequency scaling factors are empirically determined through comparison with experimental data. This approach delivers IR spectra with accuracy approaching high-level DFT methods at computational speeds comparable to semi-empirical GFN2-xTB calculations [24].
The MLatom software package provides comprehensive tools for AIQM-based IR spectrum simulation, including tutorials and scripts for calculating spectra, visualizing vibrational normal modes, and assigning bands to specific molecular vibrations [58].
Protocol for IR Spectrum Simulation
Step 1: Molecular Structure Preparation
- Obtain molecular structure from experimental data or preliminary geometry optimization
- Ensure appropriate initial geometry for the molecular state of interest (ground state, excited state)
Step 2: Geometry Optimization
- Use AIQM1 or AIQM2 method for geometry optimization
- Employ default convergence criteria or tighten for sensitive systems
- Verify optimization success through frequency analysis (no imaginary frequencies for minima)
Step 3: Harmonic Frequency Calculation
- Compute analytical Hessian matrix using AIQM method
- For AIQM2: Sum numerical Hessian from GFN2-xTB*, analytical NN Hessian, and numerical D4 dispersion Hessian
- Solve mass-weighted Hessian eigenvalue problem to obtain harmonic frequencies
- Calculate IR intensities from dipole moment derivatives
Step 4: Frequency Scaling and Spectrum Generation
- Apply empirical scaling factors: 0.95-0.98 for AIQM1, 0.96-0.99 for AIQM2 (system-dependent)
- Generate stick spectrum with calculated frequencies and intensities
- Apply line broadening functions (Lorentzian/Gaussian) for realistic spectrum simulation
Step 5: Spectrum Interpretation
- Use visualization tools to animate normal modes for frequency assignment
- Correlate calculated frequencies with experimental reference data
- Identify characteristic functional group vibrations for structural elucidation
For anharmonic spectra, alternative approaches include molecular dynamics simulations with AIQM potentials or vibrational perturbation theory (VPT2) implementations, both supported in the MLatom package [24].
Research Reagent Solutions
Table 3: Essential Computational Tools for AIQM-Based Research
| Tool Name | Type | Function | Availability |
|---|---|---|---|
| MLatom | Software Package | Primary interface for AIQM calculations | https://github.com/dralgroup/mlatom [26] |
| UAIQM | Model Library | Hosts AIQM1, AIQM2, and related models | Part of MLatom package [26] |
| xTB | Software | GFN-xTB methods used as AIQM2 baseline | https://github.com/grimme-lab/xtb [101] |
| CREST | Software | Conformational sampling for complex systems | Part of xTB package [101] |
| ANI-1x/ANI-1ccx | Dataset | Training data for AIQM models | Reference data [97] |
| QUID | Benchmark Set | Non-covalent interactions for drug-like molecules | Reference data [100] |
AIQM1 and AIQM2 represent transformative advancements in computational chemistry, effectively bridging the long-standing gap between accuracy and computational efficiency in quantum chemical simulations. By synergistically combining semiempirical quantum mechanical methods with neural network corrections and dispersion treatments, these approaches achieve CCSD(T)-level accuracy at semiempirical computational costs.
The application of these methods to IR spectroscopy demonstrates their practical utility in chemical research, enabling accurate prediction of molecular vibrational spectra for systems where high-level quantum chemical calculations were previously prohibitive. As these methods continue to evolve and integrate with emerging computational architectures, they hold the potential to dramatically accelerate research across chemistry, materials science, and drug discovery.
For researchers engaged in IR spectra simulation, AIQM methods offer a compelling alternative to traditional DFT approaches, providing superior accuracy in many cases while significantly reducing computational resource requirements. The availability of these methods in open-source software packages like MLatom ensures broad accessibility to the scientific community, promising to accelerate adoption and further development of AI-enhanced quantum mechanical methods.
The accurate analysis of complex chemical mixtures presents a significant challenge in fields ranging from materials science to pharmaceutical development. Traditional analytical techniques, such as gas chromatography/mass spectrometry (GC/MS), can be cumbersome for discriminating between highly similar complex mixtures, as they are sensitive to a limited number of components relative to the extremely large number present in many real-world samples [102]. Furthermore, for functionalized materials like graphene quantum dots (GQDs), identifying the presence and position of functional groups using standard Infrared (IR) spectroscopy is difficult because the technique traditionally assumes the samples are three-dimensional (3D) materials [103].
Sensor arrays, inspired by the combinatorial nature of biological olfactory systems, offer a powerful alternative by treating the mixture as a single analyte and generating a composite response [102] [104]. When combined with physical modeling and semi-empirical computational methods, these approaches enable the quantitative prediction of mixture components and provide insight into the molecular structure of complex materials. This Application Note details protocols for leveraging semi-empirical IR simulation and physical sensor array modeling to analyze complex mixtures, with a specific focus on functionalized nanomaterials.
Theoretical Background
The Challenge of Complex Mixtures and 2D Materials
Complex mixtures like coffee aromas can contain over 1000 discrete chemical compounds, making discrimination among similar mixtures a formidable task [102]. Similarly, for 2D carbon materials like graphene quantum dots (GQDs), conventional analytical techniques struggle to identify whether functional groups are located at the edge or the center of the 2D surface [103]. This determination is crucial, as functionalization location can drastically alter the material's electronic properties and chemical behavior [103].
Semi-Empirical Methods for IR Spectral Simulation
Semi-empirical computational methods offer a simpler and faster approach for simulating infrared (IR) spectra compared to more complex ab initio methods. These methods, such as the Austin Model 1 (AM1), have proven successful in simulating the vibrational spectrum of organic materials and agree well with experimental results [103]. They function by parameterizing the Schrödinger equation, significantly reducing computational cost while maintaining accuracy for predicting vibrational modes and IR intensities.
Physical Modeling of Sensor Arrays
Combinatorial sensor arrays utilize multiple sensors with partially overlapping specificities to respond to a given analyte or mixture. A physical model that explicitly accounts for receptor-ligand interactions can decode the array's output to predict component concentrations. The signal from a sensor is often proportional to the probability of receptor-ligand binding, incorporating parameters such as binding affinity and efficacy (ranging from 1 for a full agonist to 0 for a full antagonist) [104]. Bayesian inference is then frequently employed to analyze the output and quantify mixture constituents, even for highly related compounds [104].
Application Notes & Experimental Protocols
Protocol 1: Semi-Empirical IR Simulation for Functionalized GQDs
This protocol outlines the steps to simulate and analyze the IR spectra of pyrene-like graphene quantum dot (GQD) models to identify functional group location [103].
Research Reagent Solutions & Computational Tools
Table 1: Key Research Reagents and Computational Tools for IR Simulation
| Item Name | Function/Description |
|---|---|
| Winmostar Package | Software package containing the AM1 semi-empirical method for geometry optimization and vibrational frequency calculation [103]. |
| ZINDO Method | Zerner's Intermediate Neglect of Differential Overlap method for calculating excitation spectra and electronic transitions (e.g., HOMO-LUMO) [103]. |
| Pyrene-like GQD (pGQD) | A molecular model, such as a 9 or 16-ring structure, used as a basis for simulating functionalized GQDs [103]. |
| Functionalized GQD (fGQD) | A pGQD model functionalized with groups like hydroxyl (–OH), methyl (–CH3), or oxygen atoms at edge or center positions [103]. |
Workflow and Methodology
The following diagram illustrates the computational workflow for semi-empirical IR simulation.
Detailed Procedural Steps:
- Initial Molecular Structure Model: Build the initial geometry of the target molecule (e.g., benzene, pyrene, or a larger pyrene-like GQD) using standard bond lengths and angles. For polyaromatic hydrocarbons (PAHs), an initial carbon-carbon distance of 1.397 Å and a carbon-hydrogen distance of 1.084 Å with bond angles of 120° is a standard approximation [103].
- Geometry Optimization:
- Perform a preliminary geometry optimization using a molecular mechanics method.
- Conduct a final re-optimization using the AM1 semi-empirical method. The calculation should run until the gradient limit of the energy change reaches 0.05 kcal/mol/Ångstrom [103].
- Vibrational Mode and IR Spectrum Simulation: Once a stable structure is obtained, perform a force calculation on the optimized geometry to derive the vibrational modes and simulate the corresponding IR spectrum.
- Spectral Analysis: Focus the analysis on specific spectral regions:
- 1400–2000 cm⁻¹: Correlate vibrations in this region with C=C stretching modes of the aromatic structure.
- 3000–3500 cm⁻¹: Identify peaks associated with C–H and O–H stretching vibrations, which are critical for confirming functionalization [103].
- Excitation Spectra Calculation: Perform a single-point calculation using the ZINDO method on the AM1-optimized structure to determine the excitation spectra and analyze electronic transitions in the 200–900 nm range (UV-Vis region). This helps confirm the quantum dot characteristics of the material [103].
Protocol 2: Quantitative Analysis of Mixtures Using a Physical Sensor Array Model
This protocol describes how to use a physical model and Bayesian inference to determine the concentration of components in a complex mixture using a sensor array, based on an example with engineered GPCRs and sugar nucleotides [104].
Workflow and Methodology
The following diagram outlines the process for quantitative mixture analysis using a sensor array.
Detailed Procedural Steps:
Sensor Array Calibration:
- Experiment: For each individual ligand and each receptor in the array, measure the dose-response curve (reporter activation as a function of ligand concentration). An example system uses engineered G-protein-coupled receptors (GPCRs) in yeast strains linked to a lacZ reporter gene [104].
- Model Fitting: Use a physical model for a single receptor-ligand pair. The observed signal ((S)) is proportional to the probability that the receptor is bound, given by: ( S = \alpha + \beta \cdot \left( \frac{[L] \cdot \epsilon}{[L] + Kd} \right) ) where ([L]) is the ligand concentration, (Kd) is the dissociation constant, (\epsilon) is the efficacy (1 for agonist, 0 for antagonist), and (\alpha), (\beta) are scaling constants [104].
- Parameter Estimation: Use Bayesian inference (e.g., with nested sampling) on the single-ligand data to estimate the most likely values for the binding free energy (related to (K_d)), efficacy ((\epsilon)), and background intensity for every receptor-ligand combination [104].
Mixture Analysis:
- Application: Apply the unknown complex mixture to the calibrated sensor array.
- Response Measurement: Record the pattern of reporter activation from all sensors in the array.
- Concentration Inference: Using the previously determined single-ligand parameters and the physical model, perform Bayesian analysis on the array's combined response to predict the absolute concentration of each component in the mixture. The maximum number of components that can be successfully discriminated is generally twice the number of sensors in the array [104].
Data Presentation and Analysis
Key Spectral Data for GQD Analysis
Table 2: Key IR Spectral Regions for Analyzing Functionalized GQDs [103]
| Wavenumber Range (cm⁻¹) | Assigned Vibrational Mode | Structural Significance |
|---|---|---|
| 1400–2000 | C=C stretching | Aromatic backbone of the graphene quantum dot. |
| 3000–3500 | C–H and O–H stretching | Indicates presence of hydrogen and hydroxyl functional groups. |
Sensor Array Performance Metrics
The performance of a sensor array for mixture analysis is governed by several key principles derived from physical modeling [104]:
- Dimensionality: An effective colorimetric sensor array should have high dimensionality, potentially requiring 18 dimensions or more to define 90% of the total variance in complex mixtures like coffee aromas [102].
- Sensor Number: The maximum number of mixture components that can be successfully discriminated is theoretically up to twice the number of sensors in the array [104].
- Antagonistic Responses: The inclusion of sensors with antagonistic responses (where a ligand binds but does not activate the receptor) is essential for the accurate prediction of component concentrations in complex mixtures, as it increases the dimensionality of the response pattern [104].
- Optimal Sensor Parameters: Well-designed sensor parameters are relatively stable and a single optimized array can be useful for analyzing a wide range of mixture concentrations [104].
The integration of semi-empirical computational methods with physical modeling of sensor arrays provides a robust framework for analyzing complex chemical mixtures and functionalized nanomaterials. The protocols outlined here enable researchers to correlate the IR spectral features of 2D materials like GQDs with specific functionalization sites and to quantitatively decode the composition of complex mixtures using combinatorial sensor arrays. These methods, which leverage tools such as AM1, ZINDO, and Bayesian inference, offer a powerful toolkit for advancing research in drug development, materials science, and analytical chemistry.
Semi-empirical quantum chemical (SQC) methods provide a computationally efficient framework for calculating molecular properties by leveraging approximations and empirical parameters. The accuracy of these methods is critically evaluated against three core performance metrics: heat of formation, molecular geometry, and vibrational frequencies. These metrics are essential for validating methods intended for IR spectra simulation and other chemical applications. This note details standardized protocols for benchmarking studies and presents quantitative performance data on popular semi-empirical methods, providing researchers with a clear understanding of their capabilities and limitations in the context of spectroscopic analysis.
Performance Metrics and Benchmarking Data
The performance of semi-empirical methods is typically quantified by comparing calculated values to experimental data or high-level ab initio computational results. The key metrics include mean absolute error (MAE), root mean square error (RMSE), and maximum deviation.
Table 1: Performance Benchmark of Semi-Empirical Methods for Key Properties
| Method | Heat of Formation (kcal/mol) | Bond Length (Å) | Vibrational Frequencies (cm⁻¹) | Primary Application |
|---|---|---|---|---|
| PM6 (Default) | ~5-10 (MAE) | ~0.01-0.02 (MAE) | >70 (MAE) on benzene [105] | General organic/biological molecules |
| Reparam. PM6 (c-C3H2, C2H4, C4H6) | Data missing | Data missing | ~22 (MAE) on naphthalene [105] | Anharmonic vibrational frequencies of PAHs |
| AM1 | ~5-10 (MAE) | ~0.01-0.02 (MAE) | Successful for organic materials [3] | IR spectra of organic materials, GQDs |
| GFN-xTB | Data missing | Data missing | Varies (DFTB-type method) [20] | Geometries, frequencies, non-covalent interactions |
| DFT (e.g., B3LYP/4-31G) | Data missing | Data missing | ~12 (MAE) for scaled harmonics [105] | Reference method for PAH databases |
Table 2: Performance on Specific Molecular Systems
| Molecular System | Method | Metric | Performance | Note |
|---|---|---|---|---|
| Naphthalene (C₁₀H₈) | Reparam. PM6 [105] | Vibrational Frequency MAE | <22 cm⁻¹ | Combined with default PM6 for frequencies <3000 cm⁻¹ |
| Benzene (C₆H₆) | Reparam. PM6 [105] | Vibrational Frequency MAE | 38.7 cm⁻¹ | On test set of 5 small hydrocarbons |
| Liquid Water | PM6-fm (force-matched) [20] | Static/Dynamic Properties | Quantitative reproduction | Reparametrized for water |
| Liquid Water | Original SQC methods (AM1, PM6, DFTB2) [20] | Hydrogen Bonding | Too weak, poor property prediction | Non-reparameterized versions |
| GQDs & PAHs | AM1 [3] | IR Spectra Pattern & Intensity | Successful prediction | Useful for identifying functional groups in 2D materials |
Experimental Protocols
Protocol 1: Benchmarking Vibrational Frequencies for PAHs
This protocol outlines the procedure for reparameterizing a semi-empirical method and benchmarking its performance on polycyclic aromatic hydrocarbons (PAHs), as derived from recent research [105].
Define Training and Test Sets:
- Training Set: Select small, relevant molecules. An effective set includes cyclopropenylidene (c-C3H2), ethylene (C2H4), 1,3-butadiene (C4H6), cyclobutadiene (c-C4H2), and the cyclobutadienyl cation (c-C4H3+). These molecules contain structural motifs (e.g., aromatic rings, double bonds) found in larger PAHs.
- Test Set: Use molecules like benzene (C6H6), naphthalene (C10H8), and cyclopentadienyl anion (C5H5-) to evaluate the transferability of the optimized parameters.
Acquire Reference Data: Obtain experimental anharmonic vibrational frequencies for all molecules in the training set from reliable spectroscopic databases or literature.
Select Initial Parameters: Begin with the default parameter set of a chosen semi-empirical method (e.g., PM6). The parameters for optimization are typically the one-center, one-electron integrals (
U_SS,U_PP) and the orbital exponents (Z_S,Z_P) for the relevant elements.Compute Quartic Force Fields (QFFs): For each molecule in the training set, compute the anharmonic frequencies using a QFF approach.
- Perform automated displacements of Cartesian coordinates from the equilibrium geometry.
- For each displaced geometry, run a single-point energy calculation using the current semi-empirical parameter set.
- Fit the resulting potential energy points to a quartic (fourth-order) force field.
- Use vibrational perturbation theory at second order (VPT2) to calculate the anharmonic vibrational frequencies from the QFF.
Optimize Parameters: Use the Levenberg-Marquardt nonlinear least-squares algorithm to minimize the difference between the computed (from step 4) and experimental (from step 2) vibrational frequencies. The objective function is:
Min: Σ [ω_i(exp) - ω_i(calc)]²Iteratively adjust the selected parameters until the fit converges.Validate and Benchmark: Use the finalized, optimized parameter set to compute the vibrational frequencies for the test set molecules (e.g., naphthalene). Compare these results to their experimental frequencies to determine the method's transferability and accuracy, reporting metrics like MAE.
Protocol 2: Simulating IR Spectra for Functionalized 2D Materials
This protocol describes the use of semi-empirical methods to simulate and analyze the IR spectra of functionalized graphene quantum dots (GQDs) or similar 2D materials [3].
Model Construction: Build initial molecular structures representing the 2D material (e.g., a pyrene-like molecule as a GQD model). Create both a pristine model and functionalized models with specific groups (e.g., -OH, -CH3) attached at the edge or center of the surface.
Geometry Optimization:
- Initial Optimization: Perform a preliminary geometry optimization using a molecular mechanics (MM) force field to relieve severe steric clashes.
- Semi-Empirical Optimization: Use the AM1 or other semi-empirical method for final, precise geometry optimization. The calculation is considered converged when the energy gradient falls below a threshold, typically 0.05 kcal/(mol·Å). This yields a stable structure at a local minimum on the potential energy surface.
Vibrational Frequency Calculation: Using the optimized geometry, request a normal mode analysis. The software will calculate the Hessian matrix (second derivatives of energy with respect to atomic coordinates) and diagonalize it to obtain the vibrational frequencies and their corresponding IR intensities [106]. This step also confirms the geometry is at a minimum (no significant imaginary frequencies).
Spectral Analysis: Analyze the output, which lists all harmonic frequencies and their IR intensities. Compare the spectra of pristine and functionalized models. Focus on characteristic regions:
- 1400–2000 cm⁻¹: Aromatic C=C stretching modes.
- 3000–3500 cm⁻¹: C-H and O-H stretching modes.
- Identify new peaks or shifts induced by the functional groups.
Electronic Spectra (Optional): To correlate vibrational properties with electronic transitions, perform a single-point excited state calculation on the optimized geometry using the ZINDO method to simulate UV-Vis spectra [3].
Workflow and Pathway Visualizations
Reparameterization and Benchmarking Workflow
The following diagram illustrates the computational pathway for developing and validating a reparameterized semi-empirical method.
IR Spectrum Simulation Protocol
This workflow details the sequential steps for simulating and analyzing the IR spectrum of a molecule using a semi-empirical method.
The Scientist's Toolkit
Table 3: Essential Computational Tools for Semi-Empirical IR Simulations
| Tool Name | Type/Category | Function in Research |
|---|---|---|
| MOPAC / AMPAC | Software | Implementation of NDDO-type methods (AM1, PM3, PM6, PM7) for geometry optimization and frequency calculations. [30] |
| GFN-xTB | Software | DFTB-type method for fast calculation of geometries, vibrational frequencies, and non-covalent interactions. [20] [30] |
| Gaussian | Software | A leading computational chemistry package that can perform semi-empirical, ab initio, and DFT calculations for vibrational frequencies and IR intensities. [88] |
| Winmostar | Software | A computational chemistry package that includes AM1 and ZINDO methods for geometry optimization, vibrational analysis, and excitation spectra. [3] |
| CP2K | Software | A molecular simulation software that can perform semi-empirical and DFTB calculations, often used for condensed-phase systems. [30] |
| Quartic Force Field (QFF) | Methodology | A fourth-order Taylor series expansion of the potential energy surface used with VPT2 to compute explicit anharmonic frequencies. [105] |
| Vibrational Perturbation Theory (VPT2) | Methodology | A theoretical framework used to calculate anharmonic vibrational frequencies and account for resonance effects from a QFF. [107] [105] |
| ZINDO | Method | A semi-empirical method specifically parameterized for calculating electronic excitation spectra and properties of transition metal complexes. [3] [30] |
Conclusion
Semi-empirical methods for IR simulation represent a powerful balance between computational efficiency and chemical accuracy, particularly valuable for drug discovery applications involving tautomers, protonation states, and functionalized nanomaterials. The evolution from traditional methods like AM1 and PM3 to modern parameterizations like PM6 and PM7 has substantially improved reliability for biomedical systems, while emerging machine-learning potentials like AIQM and QDπ bridge the accuracy gap toward CCSD(T) quality. Future directions point toward increasingly universal force fields capable of reliably modeling diverse drug-like molecules, nucleic acid systems, and complex biological interactions. For biomedical researchers, these advancements enable more confident interpretation of experimental IR data, accelerated characterization of novel compounds, and deeper insights into molecular interactions underlying therapeutic mechanisms—ultimately enhancing drug design and materials development pipelines through more accessible computational spectroscopy.
References
- 1. An improved parameterization procedure for NDDO-descendant semi-empirical methods | Journal of Molecular Modeling [https://link.springer.com/article/10.1007/s00894-023-05499-3]
- 2. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3536963/]
- 3. Semi-empirical infrared spectra simulation of pyrene-like ... [https://www.nature.com/articles/s41598-023-29486-z]
- 4. Semi-empirical infrared spectra simulations of metal ... [https://www.sciencedirect.com/science/article/abs/pii/S0022286003002011]
- 5. Semiempirical Quantum Mechanical Methods for Noncovalent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4867870/]
- 6. The Accuracy of Semi-Empirical Quantum Chemistry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9657144/]
- 7. Optimization of parameters for semiempirical methods VI ... [https://link.springer.com/article/10.1007/s00894-012-1667-x]
- 8. Sparkle/PM7 Lanthanide Parameters for the Modeling of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3806451/]
- 9. Benchmark of Semiempirical Methods - Dral's Group [http://dr-dral.com/benchmark-of-semiempirical-methods/]
- 10. Influence of the computational methods in ... [https://www.sciencedirect.com/science/article/pii/S0924203125000979]
- 11. Austin Model 1 [https://en.wikipedia.org/wiki/Austin_Model_1]
- 12. Semiempirical Quantum Chemistry: Introduction. [https://people.chem.ucsb.edu/kahn/kalju/chem226/public/semiemp_intro.html]
- 13. Semi-Empirical Methods [https://gaussian.com/semiempirical/]
- 14. PM3 (chemistry) [https://en.wikipedia.org/wiki/PM3_(chemistry)]
- 15. PM6 (Parameterization Method 6) [http://www.semichem.com/ampac/pm6.php]
- 16. Performance of semiempirical DFT methods for the supramolecular... [https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp02879e]
- 17. Accurate and efficient evaluation of the ionization potentials of extreme... [https://link.springer.com/article/10.1007/s44275-024-00002-3]
- 18. Semiempirical Methods [https://www.cup.uni-muenchen.de/ch/compchem/energy/semi1.html]
- 19. Understanding Semi-Empirical Methods in Chemistry [https://www.ai-futureschool.com/en/chemistry/understanding-semi-empirical-methods-in-chemistry.php]
- 20. Benchmarking semi-empirical quantum chemical methods ... [https://arxiv.org/html/2503.11867v1]
- 21. A Dataset of Raman and Infrared Spectra as an Extension ... [https://www.nature.com/articles/s41597-025-05289-x]
- 22. Ab Initio Accuracy Neural Network Potential for Drug-Like ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12377527/]
- 23. Comparison of computational chemistry methods for the ... [https://www.nature.com/articles/s41598-020-79153-w]
- 24. Accurate and Affordable Simulation of Molecular Infrared ... [https://chemrxiv.org/engage/chemrxiv/article-details/67f735ea6dde43c908314e1c]
- 25. ML-enhanced Fast and Interpretable Simulation of IR Spectra [http://dr-dral.com/ml-enhanced-fast-and-interpretable-simulation-of-ir-spectra/]
- 26. AIQM2: organic reaction simulations beyond DFT [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc02802g]
- 27. IR Spectroscopy: From Experimental Spectra to High ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12621253/]
- 28. Predicting accurate binding energies and vibrational ... [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp01151e]
- 29. Semiempirical methods - ORCA Input Library [https://sites.google.com/site/orcainputlibrary/semiempirical-methods]
- 30. Semi-empirical quantum chemistry method [https://en.wikipedia.org/wiki/Semi-empirical_quantum_chemistry_method]
- 31. User Guide to Semiempirical Tight Binding — xtb doc 2023 ... [https://xtb-docs.readthedocs.io/]
- 32. Rapid geometry optimization for semi-empirical molecular ... [https://www.sciencedirect.com/science/article/pii/0009261471802944]
- 33. Accurate and Affordable Simulation of Molecular Infrared ... [https://chemrxiv.org/engage/chemrxiv/article-details/672b3e457be152b1d02bf1d5]
- 34. PyRAMD Scheme: A Protocol for Computing the Infrared ... [https://www.mdpi.com/2813-446X/2/3/12]
- 35. Recent Advancements in Graphene Quantum Dot‐Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12457731/]
- 36. A Review of Advances in Graphene Quantum Dots [https://www.mdpi.com/2311-5629/10/1/7]
- 37. Fluorescent graphene quantum dots: Properties regulation ... [https://www.sciencedirect.com/science/article/pii/S2773045X2500007X]
- 38. Development of advanced drug-loaded graphene quantum ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d4ra07998a]
- 39. A review on the applications of graphene quantum dots ... [https://link.springer.com/article/10.1007/s44371-025-00338-1]
- 40. Graphene Quantum Dots Market (2025-2034) [https://www.usdanalytics.com/industry-reports/graphene-quantum-dots-market]
- 41. synthesis and pharmacology of Schiff base ligands with ... [https://link.springer.com/article/10.1007/s44371-025-00228-6]
- 42. Semiempirical infrared spectra simulations for some ... [https://www.sciencedirect.com/science/article/abs/pii/S1386142501004528]
- 43. Recent advances in Schiff bases and Cu(II) complexes [https://www.sciencedirect.com/science/article/abs/pii/S0162013425000893]
- 44. IR: alcohols [https://orgchemboulder.com/Spectroscopy/irtutor/alcoholsir.shtml]
- 45. 12.8: Infrared Spectra of Some Common Functional Groups [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(Morsch_et_al.)/12%3A_Structure_Determination_-_Mass_Spectrometry_and_Infrared_Spectroscopy/12.08%3A_Infrared_Spectra_of_Some_Common_Functional_Groups]
- 46. Infrared (IR) Spectroscopy: A Quick Primer On Interpreting ... [https://www.masterorganicchemistry.com/2016/11/23/quick_analysis_of_ir_spectra/]
- 47. Infrared Spectrum Characteristics and Quantification of OH ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10193429/]
- 48. - (Organic Chemistry) - Vocab, Definition... | Fiveable C C Stretching [https://fiveable.me/key-terms/organic-chem/c%E2%89%A1c-stretching]
- 49. [17] The strength of hydrogen bonding: Infrared spectroscopy [https://www.sciencedirect.com/science/article/pii/S0076687972260190]
- 50. 6.3: IR Spectrum and Characteristic Absorption Bands [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_I_(Liu)/06%3A_Structural_Identification_of_Organic_Compounds-_IR_and_NMR_Spectroscopy/6.03%3A_IR_Spectrum_and_Characteristic_Absorption_Bands]
- 51. Intensified C Vibrator and Its Potential Role in Monitoring... C Stretching [https://pubmed.ncbi.nlm.nih.gov/30848918/]
- 52. Quantum simulation of preferred tautomeric state prediction [https://www.nature.com/articles/s41534-023-00767-9]
- 53. Experimental and pKa prediction aspects of tautomerism of ... [https://www.sciencedirect.com/science/article/abs/pii/S1740674917300422]
- 54. Towards the comprehensive, rapid, and accurate ... [https://pubmed.ncbi.nlm.nih.gov/20354892/]
- 55. Protoss: a holistic approach to predict tautomers and ... [https://jcheminf.biomedcentral.com/articles/10.1186/1758-2946-6-12]
- 56. Conformers, tautomers, and protonation states [https://pubmed.ncbi.nlm.nih.gov/37003741/]
- 57. MACE4IR: A foundation model for molecular infrared ... [https://arxiv.org/html/2508.19118v1]
- 58. ML-enhanced Fast and Interpretable Simulation of IR Spectra [http://mlatom.com/ml-enhanced-fast-and-interpretable-simulation-of-ir-spectra/]
- 59. Accurate and Affordable Simulation of Molecular Infrared ... [https://pubmed.ncbi.nlm.nih.gov/40223461/]
- 60. Molecular IR spectra simulations online! [http://mlatom.com/molecular-ir-spectra-simulations-online/]
- 61. chem IT Services - HyperChem Evaluation Version [https://www.chemits.com/en/software/molecular-modeling/hyperchem/hyperchem-demo.html]
- 62. HyperChem News [http://www.hypercubeusa.com/News/tabid/353/Default.aspx]
- 63. – AI-enhanced computational chemistry MLatom [http://mlatom.com/]
- 64. 8.0 HyperChem ( Download ) - chem.exe Free trial [https://hyperchem.software.informer.com/8.0/]
- 65. Mass scaling for vibrational frequencies from ab initio ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261405000370]
- 66. Optimization of parameters for semiempirical methods V [https://pmc.ncbi.nlm.nih.gov/articles/PMC2039871/]
- 67. A new computational methodology for the characterization of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11328912/]
- 68. PM3 semi-empirical IR spectra simulations for metal ... [https://www.sciencedirect.com/science/article/abs/pii/S0022286003002023]
- 69. A Transferable H-Bonding Correction for Semiempirical Quantum-Chemical Methods - PubMed [https://pubmed.ncbi.nlm.nih.gov/26614342/]
- 70. New Approach for Correcting Noncovalent Interactions in Semiempirical Quantum Mechanical Methods: The Importance of Multiple-Orientation Sampling - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8486165/]
- 71. Density functional benchmark for quadruple hydrogen bonds [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp00836k]
- 72. Infrared spectroscopic study of hydrogen bonding ... [https://www.nature.com/articles/s41467-020-19226-6]
- 73. 8.2: Types of Transition and Selection Rules for Transition ... [https://chem.libretexts.org/Courses/East_Tennessee_State_University/CHEM_4110%3A_Advanced_Inorganic_Chemistry/08%3A_Coordination_Chemistry_-_Spectroscopy/8.02%3A_Types_of_Transition_and_Selection_Rules_for_Transition_Metal_Complexes]
- 74. Experimental and computational studies on transition ... [https://www.sciencedirect.com/science/article/abs/pii/S0022286022015162]
- 75. Walk-through of Simulating and Fitting a Simple Spectrum [https://pgopher.chm.bris.ac.uk/Help/n2oeg.htm]
- 76. Geometry optimization — AMS 2025.1 documentation - SCM [https://www.scm.com/doc/AMS/Tasks/Geometry_Optimization.html]
- 77. Structure optimization — ASE documentation [https://wiki.fysik.dtu.dk/ase/ase/optimize.html]
- 78. 基于背景电荷计算分子在晶体环境中的吸收光谱 [http://sobereva.com/579]
- 79. gaussian - Convergence problem in geometry optimization ... [https://mattermodeling.stackexchange.com/questions/11236/convergence-problem-in-geometry-optimization-in-gaussian16]
- 80. 一种介于DFT和MD之间的模拟方法:xTB [https://www.huasuankeji.com/news/?p=348391]
- 81. 使用sobMECP程序结合Gaussian程序搜索极小能量交叉点 [http://sobereva.com/286]
- 82. Modern semiempirical electronic structure methods and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10052497/]
- 83. The Case of GFN2-xTB [https://pubmed.ncbi.nlm.nih.gov/37676972/]
- 84. Transfer-Learning Deep Raman Models Using Semiempirical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12264939/]
- 85. Real-time inline-IR-analysis via linear-combination strategy ... [https://www.nature.com/articles/s42004-025-01676-y]
- 86. Harmonic Vibrational Frequency Simulation of ... [https://www.mdpi.com/1420-3049/28/12/4638]
- 87. The application of infrared spectroscopy and DFT ... [https://www.sciencedirect.com/science/article/abs/pii/S1386142525012077]
- 88. A Dataset of Raman and Infrared Spectra as an Extension ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12137555/]
- 89. Leveraging infrared spectroscopy for automated structure ... [https://www.nature.com/articles/s42004-024-01341-w]
- 90. Setting new benchmarks in AI-driven infrared structure ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00131e]
- 91. Leveraging active learning-enhanced machine ... [https://www.nature.com/articles/s41524-025-01827-8]
- 92. Unlocking the Potential of Machine Learning in Enhancing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12079248/]
- 93. Machine learning molecular dynamics for the simulation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5636952/]
- 94. The Accuracy of Semi-Empirical Quantum Chemistry ... [https://www.mdpi.com/1422-0067/23/21/13371]
- 95. Benchmark of Approximate Quantum Chemical and Machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288007/]
- 96. A Deep Learning-Augmented Density Functional Framework ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12381711/]
- 97. Artificial intelligence-enhanced quantum chemical method ... [https://www.nature.com/articles/s41467-021-27340-2]
- 98. Advancing materials discovery through artificial intelligence [https://www.sciencedirect.com/science/article/pii/S2352940725003981]
- 99. AIQM2: organic reaction simulations beyond DFT - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12341561/]
- 100. Extending quantum-mechanical benchmark accuracy to ... [https://www.nature.com/articles/s41467-025-63587-9]
- 101. Performance of semiempirical DFT methods for the ... [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp02879e]
- 102. Discrimination of Complex Mixtures by a Colorimetric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2947826/]
- 103. - Semi of pyrene-like molecules... empirical infrared spectra simulation [https://www.nature.com/articles/s41598-023-29486-z?error=cookies_not_supported]
- 104. Decoding Complex Chemical Mixtures with a Physical Model ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1002224]
- 105. Taming semi-empirical methods for PAHs and vibrational ... [https://www.sciencedirect.com/science/article/abs/pii/S002228522300111X]
- 106. Vibrational Spectroscopy — AMS 2025.1 documentation - SCM [https://www.scm.com/doc/AMS/Vibrational_Spectroscopy.html]
- 107. Optimized Structure and Vibrational Properties by Error ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3787481/]