Research Articles
DFT vs Wavefunction Theory: The Ultimate Guide to Cost-Accuracy Trade-offs in Computational Drug Discovery
This article provides a comprehensive comparison of Density Functional Theory (DFT) and wavefunction-based electronic structure methods, focusing on the critical balance between computational cost and accuracy for researchers and drug...
Optimizing Computational Parameters for Enhanced Prediction Accuracy in Scientific Machine Learning
This article provides a comprehensive guide for researchers and drug development professionals on optimizing computational parameters to maximize prediction accuracy in scientific models.
A Practical Guide to Troubleshooting SCF Convergence in Computational Chemistry
Self-Consistent Field (SCF) convergence is a fundamental yet often challenging step in computational chemistry calculations, directly impacting the reliability of results in drug design and materials science.

Building Confidence in AI: A Robust Workflow for Validating Molecular Property Predictions
Accurate prediction of molecular properties is crucial for accelerating drug discovery and materials science, yet models trained on limited, biased, or inconsistent data can produce misleading results.
Statistical Methods and Machine Learning: Revolutionizing Computational Chemistry in Drug Discovery
This article explores the transformative role of statistical techniques and machine learning in modern computational chemistry, with a specific focus on accelerating drug discovery.
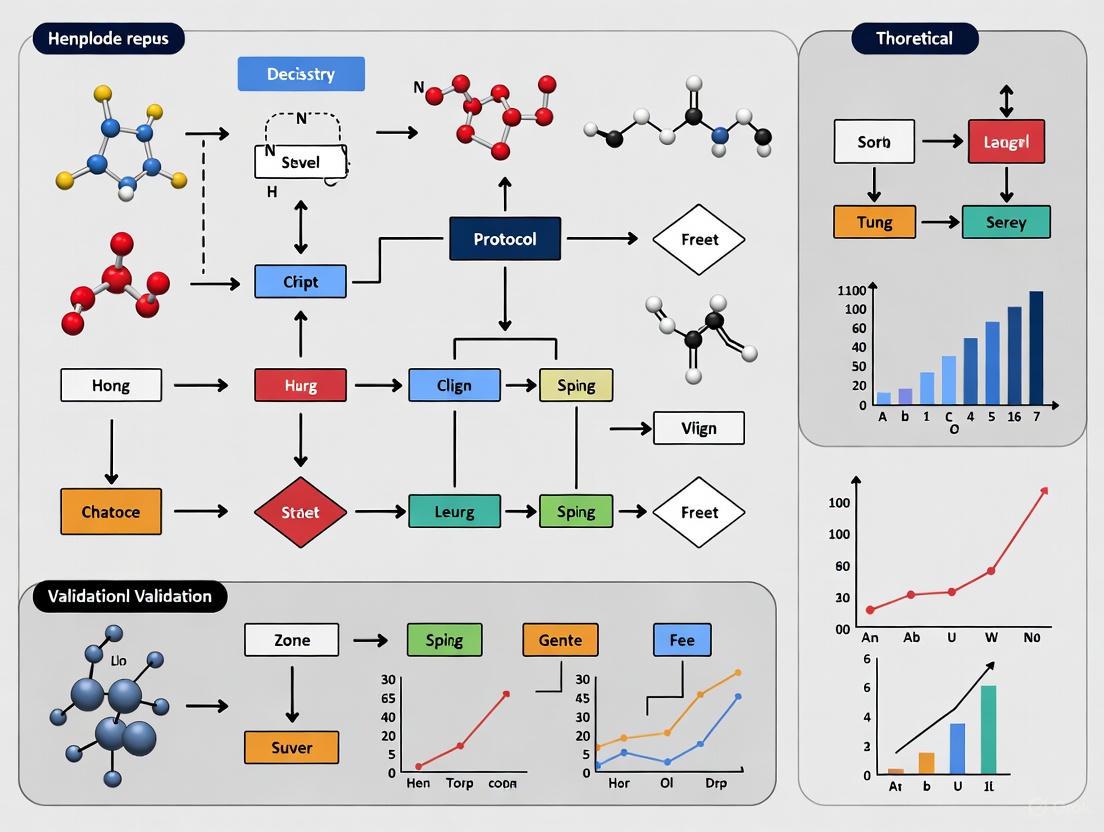
High-Throughput Validation of Chemical Probes: A Comprehensive Protocol for Robust Assay Development and Target Engagement
This article provides a detailed protocol for the high-throughput validation of chemical probes, essential tools for drug discovery and basic research.
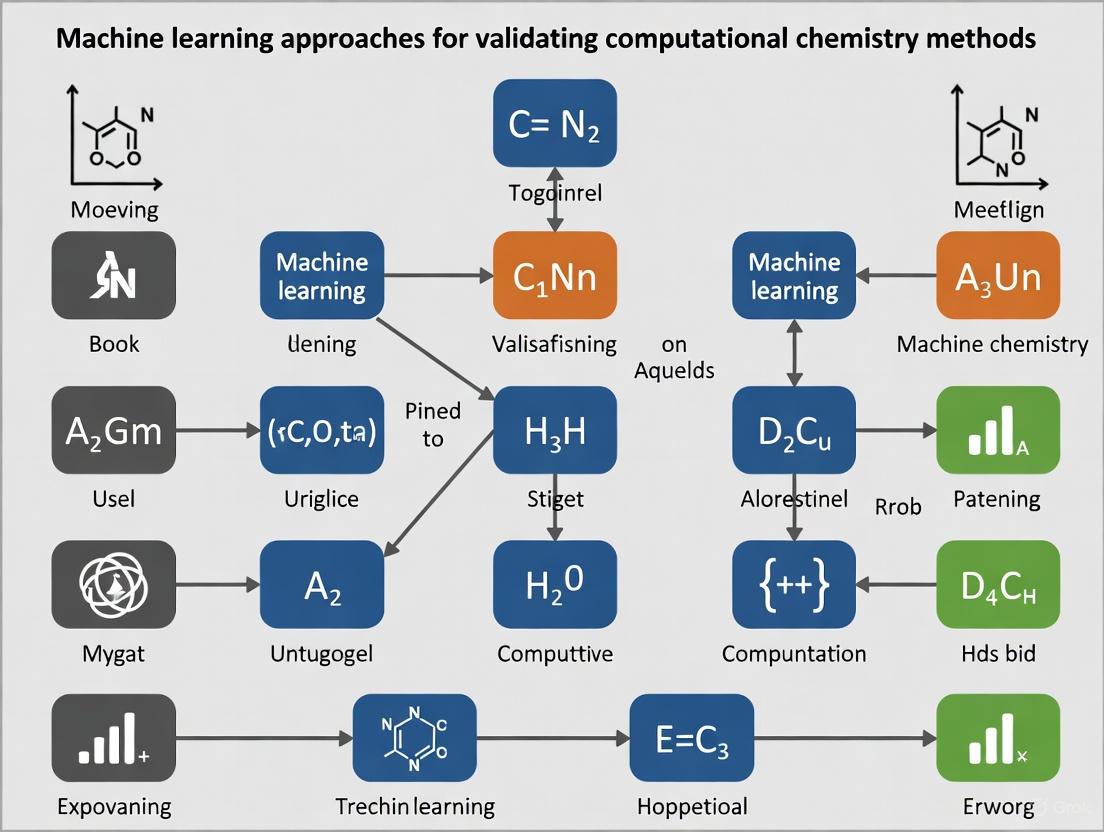
Validating the Future: A Comprehensive Guide to Machine Learning Approaches in Computational Chemistry
This article provides a comprehensive overview of machine learning (ML) validation frameworks within computational chemistry, tailored for researchers and drug development professionals.
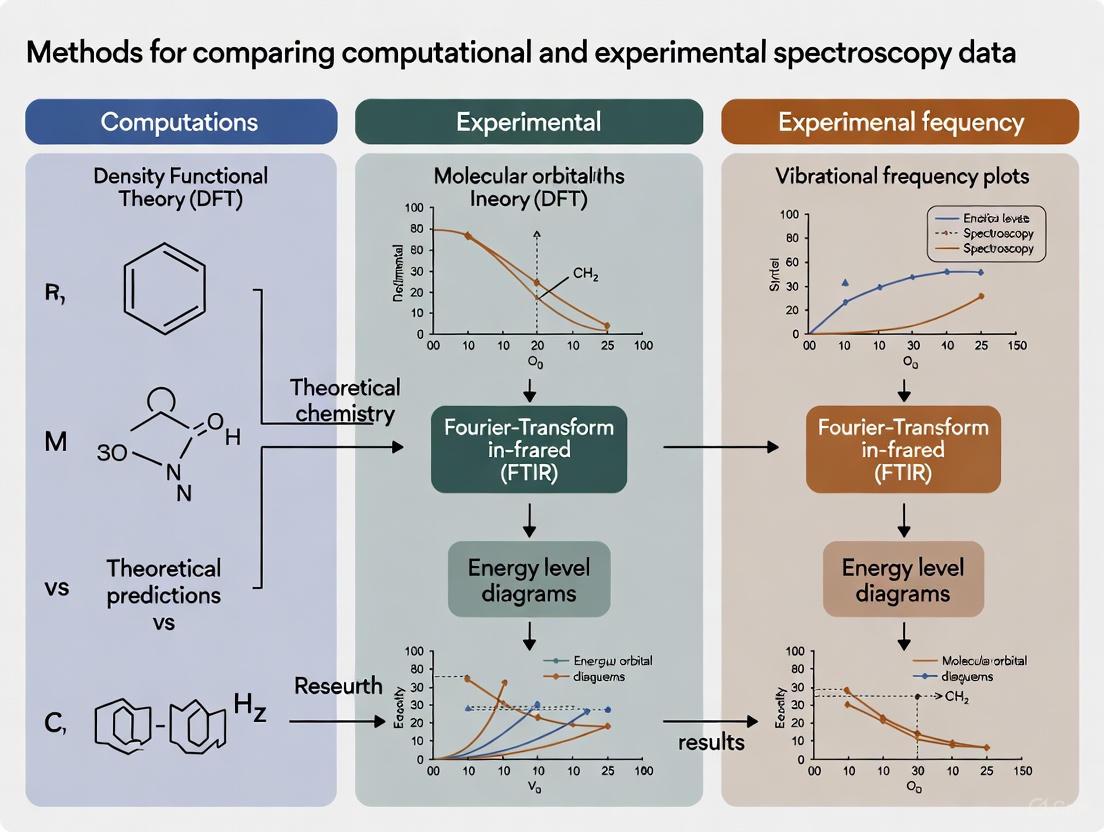
Bridging the Gap: Machine Learning Methods for Comparing Computational and Experimental Spectroscopy Data
This article explores the transformative role of machine learning (ML) in bridging computational and experimental spectroscopy, a critical synergy for researchers in chemistry, materials science, and drug development.
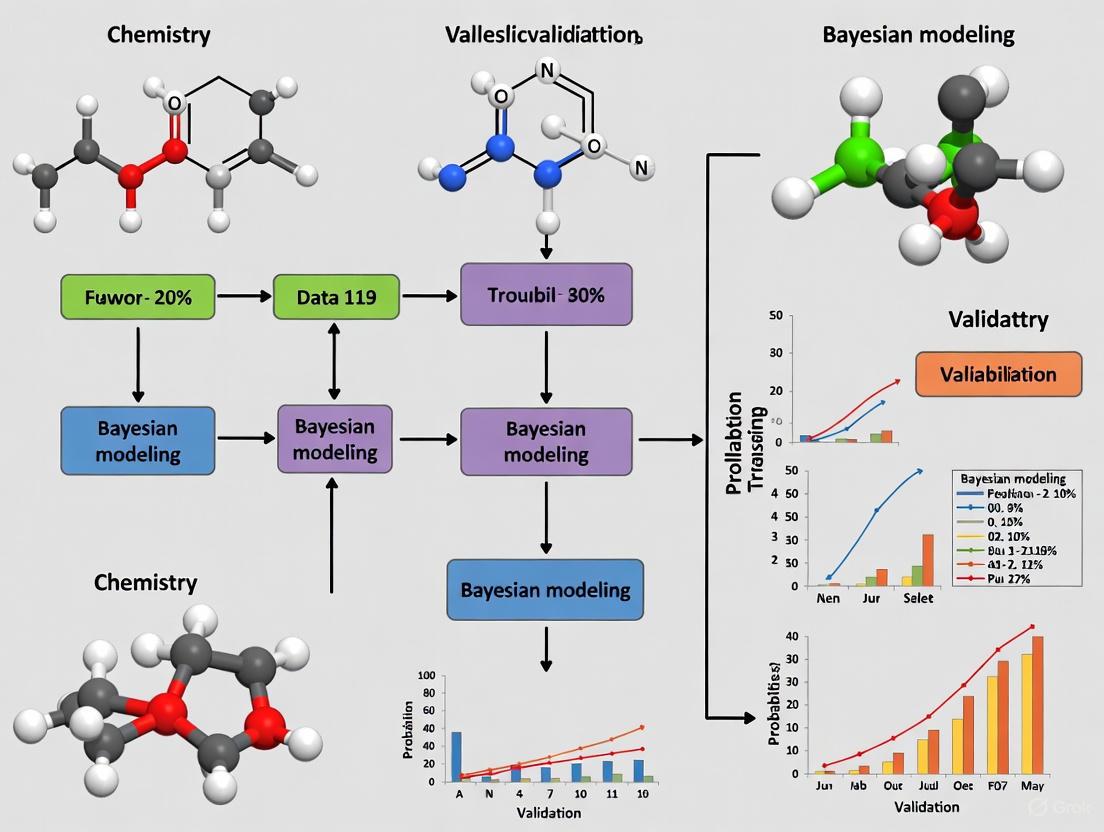
From Lab to License: A Practical Guide to Bayesian Models in Chemistry and Drug Development Validation
This article provides a comprehensive guide for researchers and drug development professionals on the practical application of Bayesian statistical models in chemistry validation.
Validating Density Functional Theory: A Practical Guide for Biomedical Researchers
Density Functional Theory (DFT) is a cornerstone of computational materials science and drug discovery, but its predictive power hinges on rigorous validation.